
Development of CHARMM-Compatible Force-Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations
Abstract
Corrinoid cofactors such as cobalamin are used by many enzymes and are essential for most living organisms. Therefore, there is broad interest in investigating cobalamin–protein interactions with molecular dynamics simulations. Previously developed parameters for cobalamins are based mainly on crystal structure data. Here, we report CHARMM-compatible force field parameters for several corrinoids developed from quantum mechanical calculations. We provide parameters for corrinoids in three oxidation states, Co3+, Co2+, and Co1+, and with various axial ligands. Lennard-Jones parameters for the cobalt center in the Co(II) and Co(I) states were optimized using a helium atom probe, and partial atomic charges were obtained with a combination of natural population analysis (NPA) and restrained electrostatic potential (RESP) fitting approaches. The Force Field Toolkit was used to optimize all bonded terms. The resulting parameters, determined solely from calculations of cobalamin alone or in water, were then validated by assessing their agreement with density functional theory geometries and by analyzing molecular dynamics simulation trajectories of several corrinoid proteins for which X-ray crystal structures are available. In each case, we obtained excellent agreement with the reference data. In comparison to previous CHARMM-compatible parameters for cobalamin, we observe a better agreement for the fold angle and lower RMSDmore »
- Authors:
-
 [2];
[1]
[2];
[1]
- Georgia Inst. of Technology, Atlanta, GA (United States)
- Oak Ridge National Lab. (ORNL), Oak Ridge, TN (United States)
- Publication Date:
- Research Org.:
- Oak Ridge National Lab. (ORNL), Oak Ridge, TN (United States); Lawrence Berkeley National Lab. (LBNL), Berkeley, CA (United States). National Energy Research Scientific Computing Center (NERSC)
- Sponsoring Org.:
- USDOE Office of Science (SC), Biological and Environmental Research (BER)
- OSTI Identifier:
- 1459308
- Alternate Identifier(s):
- OSTI ID: 1480247
- Grant/Contract Number:
- AC05-00OR22725; AC02-05CH11231
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Journal of Chemical Theory and Computation
- Additional Journal Information:
- Journal Volume: 14; Journal Issue: 2; Journal ID: ISSN 1549-9618
- Publisher:
- American Chemical Society
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 37 INORGANIC, ORGANIC, PHYSICAL, AND ANALYTICAL CHEMISTRY
Citation Formats
Pavlova, Anna, Parks, Jerry M., and Gumbart, James C. Development of CHARMM-Compatible Force-Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations. United States: N. p., 2018.
Web. doi:10.1021/acs.jctc.7b01236.
Pavlova, Anna, Parks, Jerry M., & Gumbart, James C. Development of CHARMM-Compatible Force-Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations. United States. https://doi.org/10.1021/acs.jctc.7b01236
Pavlova, Anna, Parks, Jerry M., and Gumbart, James C. Mon .
"Development of CHARMM-Compatible Force-Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations". United States. https://doi.org/10.1021/acs.jctc.7b01236. https://www.osti.gov/servlets/purl/1459308.
@article{osti_1459308,
title = {Development of CHARMM-Compatible Force-Field Parameters for Cobalamin and Related Cofactors from Quantum Mechanical Calculations},
author = {Pavlova, Anna and Parks, Jerry M. and Gumbart, James C.},
abstractNote = {Corrinoid cofactors such as cobalamin are used by many enzymes and are essential for most living organisms. Therefore, there is broad interest in investigating cobalamin–protein interactions with molecular dynamics simulations. Previously developed parameters for cobalamins are based mainly on crystal structure data. Here, we report CHARMM-compatible force field parameters for several corrinoids developed from quantum mechanical calculations. We provide parameters for corrinoids in three oxidation states, Co3+, Co2+, and Co1+, and with various axial ligands. Lennard-Jones parameters for the cobalt center in the Co(II) and Co(I) states were optimized using a helium atom probe, and partial atomic charges were obtained with a combination of natural population analysis (NPA) and restrained electrostatic potential (RESP) fitting approaches. The Force Field Toolkit was used to optimize all bonded terms. The resulting parameters, determined solely from calculations of cobalamin alone or in water, were then validated by assessing their agreement with density functional theory geometries and by analyzing molecular dynamics simulation trajectories of several corrinoid proteins for which X-ray crystal structures are available. In each case, we obtained excellent agreement with the reference data. In comparison to previous CHARMM-compatible parameters for cobalamin, we observe a better agreement for the fold angle and lower RMSD in the cobalamin binding site. The approach described here is readily adaptable for developing CHARMM-compatible force-field parameters for other corrinoids or large biomolecules.},
doi = {10.1021/acs.jctc.7b01236},
journal = {Journal of Chemical Theory and Computation},
number = 2,
volume = 14,
place = {United States},
year = {Mon Jan 15 00:00:00 EST 2018},
month = {Mon Jan 15 00:00:00 EST 2018}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
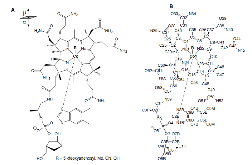 Scheme 1:: (A) Chemical structure of cobalamin. (B) Standard PDB atom numbering for cobalamin.
Scheme 1:: (A) Chemical structure of cobalamin. (B) Standard PDB atom numbering for cobalamin.
Works referenced in this record:
The Many Faces of Vitamin B12: Catalysis by Cobalamin-Dependent Enzymes
journal, June 2003
- Banerjee, Ruma; Ragsdale, Stephen W.
- Annual Review of Biochemistry, Vol. 72, Issue 1, p. 209-247
Chemistry and Enzymology of Vitamin B 12
journal, June 2005
- Brown, Kenneth L.
- Chemical Reviews, Vol. 105, Issue 6
The corrinoid from Methanobacterium thermoautotrophicum (Marburg strain). Spectroscopic structure analysis and identification as Cobeta-cyano-5'-hydroxybenzimidazolyl-cobamide (factor III)
journal, January 1987
- Krautler, Bernhard; Moll, Johanna; Thauer, Rudolf K.
- European Journal of Biochemistry, Vol. 162, Issue 2
Identification of specific corrinoids reveals corrinoid modification in dechlorinating microbial communities: Corrinoids in dechlorinating communities
journal, June 2014
- Men, Yujie; Seth, Erica C.; Yi, Shan
- Environmental Microbiology, Vol. 17, Issue 12
Vitamin B12-derivatives—enzyme cofactors and ligands of proteins and nucleic acids
journal, January 2011
- Gruber, Karl; Puffer, Barbara; Kräutler, Bernhard
- Chemical Society Reviews, Vol. 40, Issue 8
X-ray structural chemistry of cobalamins
journal, June 2006
- Randaccio, Lucio; Geremia, Silvano; Nardin, Giorgio
- Coordination Chemistry Reviews, Vol. 250, Issue 11-12
Molecular mechanics and molecular dynamics simulations of porphyrins, metalloporphyrins, heme proteins and cobalt corrinoids
journal, February 2002
- Marques, Helder M.; Brown, Kenneth L.
- Coordination Chemistry Reviews, Vol. 225, Issue 1-2
Structural insights into methyltransfer reactions of a corrinoid iron-sulfur protein involved in acetyl-CoA synthesis
journal, September 2006
- Svetlitchnaia, T.; Svetlitchnyi, V.; Meyer, O.
- Proceedings of the National Academy of Sciences, Vol. 103, Issue 39, p. 14331-14336
Structural basis for gene regulation by a B12-dependent photoreceptor
journal, September 2015
- Jost, Marco; Fernández-Zapata, Jésus; Polanco, María Carmen
- Nature, Vol. 526, Issue 7574
Vitamin B 12 in the spotlight again
journal, April 2017
- Bridwell-Rabb, Jennifer; Drennan, Catherine L.
- Current Opinion in Chemical Biology, Vol. 37
The Genetic Basis for Bacterial Mercury Methylation
journal, February 2013
- Parks, J. M.; Johs, A.; Podar, M.
- Science, Vol. 339, Issue 6125, p. 1332-1335
Structural Insights into the Mechanism of Four-Coordinate Cob(II)alamin Formation in the Active Site of the Salmonella enterica ATP:Co(I)rrinoid Adenosyltransferase Enzyme: Critical Role of Residues Phe91 and Trp93
journal, November 2012
- Moore, Theodore C.; Newmister, Sean A.; Rayment, Ivan
- Biochemistry, Vol. 51, Issue 48
Structural Characterization of a Human-Type Corrinoid Adenosyltransferase Confirms That Coenzyme B 12 Is Synthesized through a Four-Coordinate Intermediate † ‡
journal, May 2008
- St. Maurice, Martin; Mera, Paola; Park, Kiyoung
- Biochemistry, Vol. 47, Issue 21
Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins
journal, April 2010
- Randaccio, Lucio; Geremia, Silvano; Demitri, Nicola
- Molecules, Vol. 15, Issue 5
How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 å resolution
journal, March 1996
- Mancia, Filippo; Keep, Nicholas H.; Nakagawa, Atsushi
- Structure, Vol. 4, Issue 3
Crystal Structures of the BtuF Periplasmic-binding Protein for Vitamin B12 Suggest a Functionally Important Reduction in Protein Mobility upon Ligand Binding
journal, December 2002
- Karpowich, Nathan K.; Huang, Hector H.; Smith, Paul C.
- Journal of Biological Chemistry, Vol. 278, Issue 10
Visualizing molecular juggling within a B12-dependent methyltransferase complex
journal, March 2012
- Kung, Yan; Ando, Nozomi; Doukov, Tzanko I.
- Nature, Vol. 484, Issue 7393, p. 265-269
B12 cofactors directly stabilize an mRNA regulatory switch
journal, October 2012
- Johnson Jr, James E.; Reyes, Francis E.; Polaski, Jacob T.
- Nature, Vol. 492, Issue 7427
Design of a superior cytokine antagonist for topical ophthalmic use
journal, February 2013
- Hou, J.; Townson, S. A.; Kovalchin, J. T.
- Proceedings of the National Academy of Sciences, Vol. 110, Issue 10
Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation
journal, October 2014
- Payne, Karl A. P.; Quezada, Carolina P.; Fisher, Karl
- Nature, Vol. 517, Issue 7535
Structural basis for organohalide respiration
journal, October 2014
- Bommer, Martin; Kunze, Cindy; Fesseler, Jochen
- Science, Vol. 346, Issue 6208
A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules
journal, May 1995
- Cornell, Wendy D.; Cieplak, Piotr; Bayly, Christopher I.
- Journal of the American Chemical Society, Vol. 117, Issue 19
An all-atom empirical energy function for the simulation of nucleic acids
journal, December 1995
- MacKerell, Alexander D.; Wiorkiewicz-Kuczera, Joanna; Karplus, Martin
- Journal of the American Chemical Society, Vol. 117, Issue 48
CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields
journal, January 2009
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.
- Journal of Computational Chemistry
A fast empirical GAFF compatible partial atomic charge assignment scheme for modeling interactions of small molecules with biomolecular targets
journal, October 2010
- Mukherjee, Goutam; Patra, Niladri; Barua, Poranjyoti
- Journal of Computational Chemistry, Vol. 32, Issue 5
Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids
journal, January 1996
- Jorgensen, William L.; Maxwell, David S.; Tirado-Rives, Julian
- Journal of the American Chemical Society, Vol. 118, Issue 45
Classical force field parameters for the heme prosthetic group of cytochrome c
journal, October 2004
- Autenrieth, Felix; Tajkhorshid, Emad; Baudry, Jerome
- Journal of Computational Chemistry, Vol. 25, Issue 13
New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models
journal, June 2005
- Oda, Akifumi; Yamaotsu, Noriyuki; Hirono, Shuichi
- Journal of Computational Chemistry, Vol. 26, Issue 8
Revised CHARMM force field parameters for iron-containing cofactors of photosystem II
journal, August 2017
- Adam, Suliman; Knapp-Mohammady, Michaela; Yi, Jun
- Journal of Computational Chemistry, Vol. 39, Issue 1
Anab initio force field for the cofactors of bacterial photosynthesis
journal, January 2002
- Ceccarelli, Matteo; Procacci, Piero; Marchi, Massimo
- Journal of Computational Chemistry, Vol. 24, Issue 2
Estimation of Transition-Metal Empirical Parameters for Molecular Mechanical Force Fields
journal, July 2016
- Šebesta, Filip; Sláma, Vladislav; Melcr, Josef
- Journal of Chemical Theory and Computation, Vol. 12, Issue 8
Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF)
journal, August 2010
- Peters, Martin B.; Yang, Yue; Wang, Bing
- Journal of Chemical Theory and Computation, Vol. 6, Issue 9
Automated Parametrization of AMBER Force Field Terms from Vibrational Analysis with a Focus on Functionalizing Dinuclear Zinc(II) Scaffolds
journal, January 2012
- Burger, Steven K.; Lacasse, Mike; Verstraelen, Toon
- Journal of Chemical Theory and Computation, Vol. 8, Issue 2
VFFDT: A New Software for Preparing AMBER Force Field Parameters for Metal-Containing Molecular Systems
journal, March 2016
- Zheng, Suqing; Tang, Qing; He, Jian
- Journal of Chemical Information and Modeling, Vol. 56, Issue 4
Rapid parameterization of small molecules using the force field toolkit
journal, September 2013
- Mayne, Christopher G.; Saam, Jan; Schulten, Klaus
- Journal of Computational Chemistry, Vol. 34, Issue 32
Force field design for metalloproteins
journal, October 1991
- Hoops, Stephen C.; Anderson, Kenneth W.; Merz, Kenneth M.
- Journal of the American Chemical Society, Vol. 113, Issue 22
Zinc binding in proteins and solution: A simple but accurate nonbonded representation
journal, September 1995
- Stote, Roland H.; Karplus, Martin
- Proteins: Structure, Function, and Genetics, Vol. 23, Issue 1
Comparison of Methods to Obtain Force-Field Parameters for Metal Sites
journal, July 2011
- Hu, LiHong; Ryde, Ulf
- Journal of Chemical Theory and Computation, Vol. 7, Issue 8
Novel Zinc Protein Molecular Dynamics Simulations: Steps Toward Antiangiogenesis for Cancer Treatment
journal, October 1999
- Pang, Yuan-Ping
- Journal of Molecular Modeling, Vol. 5, Issue 10
Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions
journal, January 2010
- Singh, Nidhi; Warshel, Arieh
- Proteins: Structure, Function, and Bioinformatics
Computation of structures and properties of transition metal compounds
journal, March 2009
- Comba, Peter; Kerscher, Marion
- Coordination Chemistry Reviews, Vol. 253, Issue 5-6
Robustness in the fitting of molecular mechanics parameters
journal, March 2015
- Vanommeslaeghe, Kenno; Yang, Mingjun; MacKerell, Alexander D.
- Journal of Computational Chemistry, Vol. 36, Issue 14
Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms
journal, December 1977
- Allinger, Norman L.
- Journal of the American Chemical Society, Vol. 99, Issue 25
A molecular mechanics force field for the cobalt corrinoids
journal, September 1995
- Marques, Helder M.; Brown, Kenneth L.
- Journal of Molecular Structure: THEOCHEM, Vol. 340, Issue 1-3
Parameters for the amber force field for the molecular mechanics modeling of the cobalt corrinoids
journal, April 2001
- Marques, H. M.; Ngoma, B.; Egan, T. J.
- Journal of Molecular Structure, Vol. 561, Issue 1-3
Coupling of Calcium and Substrate Binding through Loop Alignment in the Outer-Membrane Transporter BtuB
journal, November 2009
- Gumbart, James; Wiener, Michael C.; Tajkhorshid, Emad
- Journal of Molecular Biology, Vol. 393, Issue 5
Solution structure of cyanocobalamin (vitamin B12) by NMR-restrained molecular dynamics and simulated annealing calculations
journal, January 1996
- Marques, Helder M.; Hicks, Ricky P.; Brown, Kenneth L.
- Chemical Communications, Issue 12
The solution structure of some cobalamins determined by NMR-restrained molecular modelling
journal, March 2005
- Perry, Christopher B.; Brown, Kenneth L.; Zou, Xiang
- Journal of Molecular Structure, Vol. 737, Issue 2-3
Parametrization of macrolide antibiotics using the force field toolkit
journal, August 2015
- Pavlova, Anna; Gumbart, James C.
- Journal of Computational Chemistry, Vol. 36, Issue 27
Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium
journal, December 2011
- Hart, Katarina; Foloppe, Nicolas; Baker, Christopher M.
- Journal of Chemical Theory and Computation, Vol. 8, Issue 1
Correlated wavefunction methods in bioinorganic chemistry
journal, May 2011
- Neese, Frank; Liakos, Dimitrios G.; Ye, Shengfa
- JBIC Journal of Biological Inorganic Chemistry, Vol. 16, Issue 6
Density-functional exchange-energy approximation with correct asymptotic behavior
journal, September 1988
- Becke, A. D.
- Physical Review A, Vol. 38, Issue 6
Density-functional approximation for the correlation energy of the inhomogeneous electron gas
journal, June 1986
- Perdew, John P.
- Physical Review B, Vol. 33, Issue 12
Self-interaction correction to density-functional approximations for many-electron systems
journal, May 1981
- Perdew, J. P.; Zunger, Alex
- Physical Review B, Vol. 23, Issue 10, p. 5048-5079
Theoretical Prediction of the Co−C Bond Strength in Cobalamins
journal, September 2003
- Jensen, Kasper P.; Ryde, Ulf
- The Journal of Physical Chemistry A, Vol. 107, Issue 38
Structure−Energy Relations in Methylcobalamin with and without Bound Axial Base
journal, October 2004
- Rovira, Carme; Biarnés, Xevi; Kunc, Karel
- Inorganic Chemistry, Vol. 43, Issue 21
Axial Bonding in Alkylcobalamins: DFT Analysis of the Inverse Versus Normal Trans Influence
journal, July 2009
- Kuta, Jadwiga; Wuerges, Jochen; Randaccio, Lucio
- The Journal of Physical Chemistry A, Vol. 113, Issue 43
The Cobalt–Methyl Bond Dissociation in Methylcobalamin: New Benchmark Analysis Based on Density Functional Theory and Completely Renormalized Coupled-Cluster Calculations
journal, May 2012
- Kozlowski, Pawel M.; Kumar, Manoj; Piecuch, Piotr
- Journal of Chemical Theory and Computation, Vol. 8, Issue 6
Co–C Dissociation of Adenosylcobalamin (Coenzyme B 12 ): Role of Dispersion, Induction Effects, Solvent Polarity, and Relativistic and Thermal Corrections
journal, August 2014
- Kepp, Kasper P.
- The Journal of Physical Chemistry A, Vol. 118, Issue 34
Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy
journal, January 2005
- Weigend, Florian; Ahlrichs, Reinhart
- Physical Chemistry Chemical Physics, Vol. 7, Issue 18, p. 3297-3305
Accurate Coulomb-fitting basis sets for H to Rn
journal, January 2006
- Weigend, Florian
- Physical Chemistry Chemical Physics, Vol. 8, Issue 9
Toward Quantitatively Accurate Calculation of the Redox-Associated Acid–Base and Ligand Binding Equilibria of Aquacobalamin
journal, July 2016
- Johnston, Ryne C.; Zhou, Jing; Smith, Jeremy C.
- The Journal of Physical Chemistry B, Vol. 120, Issue 30
Combinedab initio/empirical approach for optimization of Lennard-Jones parameters for polar-neutral compounds
journal, December 2001
- Chen, I. Jen; Yin, Daxu; MacKerell, Alexander D.
- Journal of Computational Chemistry, Vol. 23, Issue 2
Generalized Gradient Approximation Made Simple
journal, October 1996
- Perdew, John P.; Burke, Kieron; Ernzerhof, Matthias
- Physical Review Letters, Vol. 77, Issue 18, p. 3865-3868
Density‐functional thermochemistry. III. The role of exact exchange
journal, April 1993
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 98, Issue 7, p. 5648-5652
Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density
journal, January 1988
- Lee, Chengteh; Yang, Weitao; Parr, Robert G.
- Physical Review B, Vol. 37, Issue 2
A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu
journal, April 2010
- Grimme, Stefan; Antony, Jens; Ehrlich, Stephan
- The Journal of Chemical Physics, Vol. 132, Issue 15
Effect of the damping function in dispersion corrected density functional theory
journal, March 2011
- Grimme, Stefan; Ehrlich, Stephan; Goerigk, Lars
- Journal of Computational Chemistry, Vol. 32, Issue 7
VMD: Visual molecular dynamics
journal, February 1996
- Humphrey, William; Dalke, Andrew; Schulten, Klaus
- Journal of Molecular Graphics, Vol. 14, Issue 1
A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model
journal, October 1993
- Bayly, Christopher I.; Cieplak, Piotr; Cornell, Wendy
- The Journal of Physical Chemistry, Vol. 97, Issue 40
Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint
journal, September 1988
- Reed, Alan E.; Curtiss, Larry A.; Weinhold, Frank
- Chemical Reviews, Vol. 88, Issue 6
Derivation of net atomic charges from molecular electrostatic potentials
journal, April 1990
- Woods, Robert J.; Khalil, Maged; Pell, Wendy
- Journal of Computational Chemistry, Vol. 11, Issue 3
Conformational dependence of electrostatic potential derived charges of a lipid headgroup: Glycerylphosphorylcholine
journal, June 1992
- Stouch, T. R.; Williams, Donald E.
- Journal of Computational Chemistry, Vol. 13, Issue 5
Fitting Molecular Electrostatic Potentials from Quantum Mechanical Calculations
journal, March 2007
- Hu, Hao; Lu, Zhenyu; Yang, Weitao
- Journal of Chemical Theory and Computation, Vol. 3, Issue 3
The R.E.D. tools: advances in RESP and ESP charge derivation and force field library building
journal, January 2010
- Dupradeau, François-Yves; Pigache, Adrien; Zaffran, Thomas
- Physical Chemistry Chemical Physics, Vol. 12, Issue 28
Automated conformational energy fitting for force-field development
journal, May 2008
- Guvench, Olgun; MacKerell, Alexander D.
- Journal of Molecular Modeling, Vol. 14, Issue 8
Scalable molecular dynamics with NAMD
journal, January 2005
- Phillips, James C.; Braun, Rosemary; Wang, Wei
- Journal of Computational Chemistry, Vol. 26, Issue 16, p. 1781-1802
Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles
journal, August 2012
- Best, Robert B.; Zhu, Xiao; Shim, Jihyun
- Journal of Chemical Theory and Computation, Vol. 8, Issue 9
Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types
journal, June 2010
- Klauda, Jeffery B.; Venable, Richard M.; Freites, J. Alfredo
- The Journal of Physical Chemistry B, Vol. 114, Issue 23
Comparison of simple potential functions for simulating liquid water
journal, July 1983
- Jorgensen, William L.; Chandrasekhar, Jayaraman; Madura, Jeffry D.
- The Journal of Chemical Physics, Vol. 79, Issue 2
Particle mesh Ewald: An N ⋅log( N ) method for Ewald sums in large systems
journal, June 1993
- Darden, Tom; York, Darrin; Pedersen, Lee
- The Journal of Chemical Physics, Vol. 98, Issue 12
CHARMM-GUI: A web-based graphical user interface for CHARMM
journal, March 2008
- Jo, Sunhwan; Kim, Taehoon; Iyer, Vidyashankara G.
- Journal of Computational Chemistry, Vol. 29, Issue 11
Natural population analysis
journal, July 1985
- Reed, Alan E.; Weinstock, Robert B.; Weinhold, Frank
- The Journal of Chemical Physics, Vol. 83, Issue 2
Modifying the OPLS-AA force field to improve hydration free energies for several amino acid side chains using new atomic charges and an off-plane charge model for aromatic residues
journal, January 2006
- Xu, Zhitao; Luo, Harry H.; Tieleman, D. Peter
- Journal of Computational Chemistry, Vol. 28, Issue 3
How a protein generates a catalytic radical from coenzyme B12: X-ray structure of a diol-dehydratase–adeninylpentylcobalamin complex
journal, July 2000
- Masuda, Jun; Shibata, Naoki; Morimoto, Yukio
- Structure, Vol. 8, Issue 7
Substrate-induced transmembrane signaling in the cobalamin transporter BtuB
journal, March 2003
- Chimento, David P.; Mohanty, Arun K.; Kadner, Robert J.
- Nature Structural & Molecular Biology, Vol. 10, Issue 5
Works referencing / citing this record:
Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications
journal, August 2019
- Singh, Nidhi; Li, Wenjin
- International Journal of Molecular Sciences, Vol. 20, Issue 15
Atomistic Simulations of COSAN: Amphiphiles without a Head‐and‐Tail Design Display “Head and Tail” Surfactant Behavior
journal, January 2020
- Malaspina, David C.; Viñas, Clara; Teixidor, Francesc
- Angewandte Chemie, Vol. 132, Issue 8
Atomistic Simulations of COSAN: Amphiphiles without a Head‐and‐Tail Design Display “Head and Tail” Surfactant Behavior
journal, February 2020
- Malaspina, David C.; Viñas, Clara; Teixidor, Francesc
- Angewandte Chemie International Edition, Vol. 59, Issue 8
Figures / Tables found in this record: