
Less is more: Sampling chemical space with active learning
Abstract
We present the development of accurate and transferable machine learning (ML) potentials for predicting molecular energetics is a challenging task. The process of data generation to train such ML potentials is a task neither well understood nor researched in detail. In this work, we present a fully automated approach for the generation of datasets with the intent of training universal ML potentials. It is based on the concept of active learning (AL) via Query by Committee (QBC), which uses the disagreement between an ensemble of ML potentials to infer the reliability of the ensemble’s prediction. QBC allows the presented AL algorithm to automatically sample regions of chemical space where the ML potential fails to accurately predict the potential energy. AL improves the overall fitness of ANAKIN-ME (ANI) deep learning potentials in rigorous test cases by mitigating human biases in deciding what new training data to use. AL also reduces the training set size to a fraction of the data required when using naive random sampling techniques. To provide validation of our AL approach, we develop the COmprehensive Machine-learning Potential (COMP6) benchmark (publicly available on GitHub) which contains a diverse set of organic molecules. Active learning-based ANI potentials outperform the originalmore »
- Authors:
-
 [1];
[2];
[2];
[1];
[2];
[2];
- Univ. of Florida, Gainesville, FL (United States)
- Los Alamos National Lab. (LANL), Los Alamos, NM (United States)
- Univ. of North Carolina, Chapel Hill, NC (United States)
- Publication Date:
- Research Org.:
- Los Alamos National Laboratory (LANL), Los Alamos, NM (United States)
- Sponsoring Org.:
- USDOE Office of Science (SC); USDOE National Nuclear Security Administration (NNSA)
- OSTI Identifier:
- 1479911
- Alternate Identifier(s):
- OSTI ID: 1438295
- Report Number(s):
- LA-UR-18-30171
Journal ID: ISSN 0021-9606
- Grant/Contract Number:
- AC52-06NA25396
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Journal of Chemical Physics
- Additional Journal Information:
- Journal Volume: 148; Journal Issue: 24; Journal ID: ISSN 0021-9606
- Publisher:
- American Institute of Physics (AIP)
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 37 INORGANIC, ORGANIC, PHYSICAL, AND ANALYTICAL CHEMISTRY; Material Science
Citation Formats
Smith, Justin Steven, Nebgen, Benjamin Tyler, Lubbers, Nicholas Edward, Isayev, Olexandr, and Roitberg, Adrian E. Less is more: Sampling chemical space with active learning. United States: N. p., 2018.
Web. doi:10.1063/1.5023802.
Smith, Justin Steven, Nebgen, Benjamin Tyler, Lubbers, Nicholas Edward, Isayev, Olexandr, & Roitberg, Adrian E. Less is more: Sampling chemical space with active learning. United States. https://doi.org/10.1063/1.5023802
Smith, Justin Steven, Nebgen, Benjamin Tyler, Lubbers, Nicholas Edward, Isayev, Olexandr, and Roitberg, Adrian E. Tue .
"Less is more: Sampling chemical space with active learning". United States. https://doi.org/10.1063/1.5023802. https://www.osti.gov/servlets/purl/1479911.
@article{osti_1479911,
title = {Less is more: Sampling chemical space with active learning},
author = {Smith, Justin Steven and Nebgen, Benjamin Tyler and Lubbers, Nicholas Edward and Isayev, Olexandr and Roitberg, Adrian E},
abstractNote = {We present the development of accurate and transferable machine learning (ML) potentials for predicting molecular energetics is a challenging task. The process of data generation to train such ML potentials is a task neither well understood nor researched in detail. In this work, we present a fully automated approach for the generation of datasets with the intent of training universal ML potentials. It is based on the concept of active learning (AL) via Query by Committee (QBC), which uses the disagreement between an ensemble of ML potentials to infer the reliability of the ensemble’s prediction. QBC allows the presented AL algorithm to automatically sample regions of chemical space where the ML potential fails to accurately predict the potential energy. AL improves the overall fitness of ANAKIN-ME (ANI) deep learning potentials in rigorous test cases by mitigating human biases in deciding what new training data to use. AL also reduces the training set size to a fraction of the data required when using naive random sampling techniques. To provide validation of our AL approach, we develop the COmprehensive Machine-learning Potential (COMP6) benchmark (publicly available on GitHub) which contains a diverse set of organic molecules. Active learning-based ANI potentials outperform the original random sampled ANI-1 potential with only 10% of the data, while the final active learning-based model vastly outperforms ANI-1 on the COMP6 benchmark after training to only 25% of the data. Finally, we show that our proposed AL technique develops a universal ANI potential (ANI-1x) that provides accurate energy and force predictions on the entire COMP6 benchmark. Finally, this universal ML potential achieves a level of accuracy on par with the best ML potentials for single molecules or materials, while remaining applicable to the general class of organic molecules composed of the elements CHNO.},
doi = {10.1063/1.5023802},
journal = {Journal of Chemical Physics},
number = 24,
volume = 148,
place = {United States},
year = {Tue May 22 00:00:00 EDT 2018},
month = {Tue May 22 00:00:00 EDT 2018}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
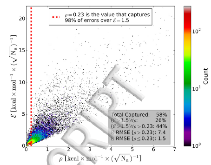 Figure 1: Example of choosing a value $\hat{ρ}$ which captures 98% of all errors ($ε$) over 1.5 kcal/mol on the GDB07to09 benchmark set using the initial (before using active learning) ANI model ensemble. The value which accomplished this is found to be $\hat{ρ}$ = 0.23. This value of $\hat{ρ}$ usedmore »
Figure 1: Example of choosing a value $\hat{ρ}$ which captures 98% of all errors ($ε$) over 1.5 kcal/mol on the GDB07to09 benchmark set using the initial (before using active learning) ANI model ensemble. The value which accomplished this is found to be $\hat{ρ}$ = 0.23. This value of $\hat{ρ}$ usedmore »
Works referenced in this record:
Neural Networks for the Prediction of Organic Chemistry Reactions
journal, October 2016
- Wei, Jennifer N.; Duvenaud, David; Aspuru-Guzik, Alán
- ACS Central Science, Vol. 2, Issue 10
Material informatics driven design and experimental validation of lead titanate as an aqueous solar photocathode
journal, October 2016
- Moot, Taylor; Isayev, Olexandr; Call, Robert W.
- Materials Discovery, Vol. 6
The S66x8 benchmark for noncovalent interactions revisited: explicitly correlated ab initio methods and density functional theory
journal, January 2016
- Brauer, Brina; Kesharwani, Manoj K.; Kozuch, Sebastian
- Physical Chemistry Chemical Physics, Vol. 18, Issue 31
Protein–Ligand Scoring with Convolutional Neural Networks
journal, April 2017
- Ragoza, Matthew; Hochuli, Joshua; Idrobo, Elisa
- Journal of Chemical Information and Modeling, Vol. 57, Issue 4
Systematic optimization of long-range corrected hybrid density functionals
journal, February 2008
- Chai, Jeng-Da; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 128, Issue 8
970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13
journal, July 2009
- Blum, Lorenz C.; Reymond, Jean-Louis
- Journal of the American Chemical Society, Vol. 131, Issue 25
Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models
journal, September 2017
- Liu, Bowen; Ramsundar, Bharath; Kawthekar, Prasad
- ACS Central Science, Vol. 3, Issue 10
UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations
journal, December 1992
- Rappe, A. K.; Casewit, C. J.; Colwell, K. S.
- Journal of the American Chemical Society, Vol. 114, Issue 25, p. 10024-10035
The EBI RDF platform: linked open data for the life sciences
journal, January 2014
- Jupp, S.; Malone, J.; Bolleman, J.
- Bioinformatics, Vol. 30, Issue 9
CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data
journal, July 2013
- Huang, Jing; MacKerell, Alexander D.
- Journal of Computational Chemistry, Vol. 34, Issue 25
A full coupled‐cluster singles and doubles model: The inclusion of disconnected triples
journal, February 1982
- Purvis, George D.; Bartlett, Rodney J.
- The Journal of Chemical Physics, Vol. 76, Issue 4
The ChEMBL bioactivity database: an update
journal, November 2013
- Bento, A. Patrícia; Gaulton, Anna; Hersey, Anne
- Nucleic Acids Research, Vol. 42, Issue D1
Virtual Exploration of the Small-Molecule Chemical Universe below 160 Daltons
journal, February 2005
- Fink, Tobias; Bruggesser, Heinz; Reymond, Jean-Louis
- Angewandte Chemie International Edition, Vol. 44, Issue 10
ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules
journal, December 2017
- Smith, Justin S.; Isayev, Olexandr; Roitberg, Adrian E.
- Scientific Data, Vol. 4, Issue 1
Structure-based sampling and self-correcting machine learning for accurate calculations of potential energy surfaces and vibrational levels
journal, June 2017
- Dral, Pavlo O.; Owens, Alec; Yurchenko, Sergei N.
- The Journal of Chemical Physics, Vol. 146, Issue 24
Self‐Consistent Molecular‐Orbital Methods. IX. An Extended Gaussian‐Type Basis for Molecular‐Orbital Studies of Organic Molecules
journal, January 1971
- Ditchfield, R.; Hehre, W. J.; Pople, J. A.
- The Journal of Chemical Physics, Vol. 54, Issue 2
Addressing uncertainty in atomistic machine learning
journal, January 2017
- Peterson, Andrew A.; Christensen, Rune; Khorshidi, Alireza
- Physical Chemistry Chemical Physics, Vol. 19, Issue 18
Virtual Exploration of the Chemical Universe up to 11 Atoms of C, N, O, F: Assembly of 26.4 Million Structures (110.9 Million Stereoisomers) and Analysis for New Ring Systems, Stereochemistry, Physicochemical Properties, Compound Classes, and Drug Discovery
journal, January 2007
- Fink, Tobias; Reymond, Jean-Louis
- Journal of Chemical Information and Modeling, Vol. 47, Issue 2
CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields
journal, January 2009
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.
- Journal of Computational Chemistry
Comparison of multiple Amber force fields and development of improved protein backbone parameters
journal, November 2006
- Hornak, Viktor; Abel, Robert; Okur, Asim
- Proteins: Structure, Function, and Bioinformatics, Vol. 65, Issue 3
Assessment of the Performance of DFT and DFT-D Methods for Describing Distance Dependence of Hydrogen-Bonded Interactions
journal, December 2010
- Thanthiriwatte, Kanchana S.; Hohenstein, Edward G.; Burns, Lori A.
- Journal of Chemical Theory and Computation, Vol. 7, Issue 1
Representing the potential-energy surface of protonated water clusters by high-dimensional neural network potentials
journal, January 2015
- Kondati Natarajan, Suresh; Morawietz, Tobias; Behler, Jörg
- Physical Chemistry Chemical Physics, Vol. 17, Issue 13
The open science grid
journal, July 2007
- Pordes, Ruth; Petravick, Don; Kramer, Bill
- Journal of Physics: Conference Series, Vol. 78
Intrinsic Bond Energies from a Bonds-in-Molecules Neural Network
journal, June 2017
- Yao, Kun; Herr, John E.; Brown, Seth N.
- The Journal of Physical Chemistry Letters, Vol. 8, Issue 12
Permutation invariant potential energy surfaces for polyatomic reactions using atomistic neural networks
journal, June 2016
- Kolb, Brian; Zhao, Bin; Li, Jun
- The Journal of Chemical Physics, Vol. 144, Issue 22
Materials Synthesis Insights from Scientific Literature via Text Extraction and Machine Learning
journal, October 2017
- Kim, Edward; Huang, Kevin; Saunders, Adam
- Chemistry of Materials, Vol. 29, Issue 21
Møller-Plesset perturbation theory: from small molecule methods to methods for thousands of atoms: Møller-Plesset perturbation theory
journal, May 2011
- Cremer, Dieter
- Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 1, Issue 4
First Principles Neural Network Potentials for Reactive Simulations of Large Molecular and Condensed Systems
journal, August 2017
- Behler, Jörg
- Angewandte Chemie International Edition, Vol. 56, Issue 42
A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu
journal, April 2010
- Grimme, Stefan; Antony, Jens; Ehrlich, Stephan
- The Journal of Chemical Physics, Vol. 132, Issue 15
Hierarchical modeling of molecular energies using a deep neural network
journal, June 2018
- Lubbers, Nicholas; Smith, Justin S.; Barros, Kipton
- The Journal of Chemical Physics, Vol. 148, Issue 24
COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase ApplicationsOverview with Details on Alkane and Benzene Compounds
journal, September 1998
- Sun, H.
- The Journal of Physical Chemistry B, Vol. 102, Issue 38
GLYCAM06: A generalizable biomolecular force field. Carbohydrates: GLYCAM06
journal, September 2007
- Kirschner, Karl N.; Yongye, Austin B.; Tschampel, Sarah M.
- Journal of Computational Chemistry, Vol. 29, Issue 4
Active-learning strategies in computer-assisted drug discovery
journal, April 2015
- Reker, Daniel; Schneider, Gisbert
- Drug Discovery Today, Vol. 20, Issue 4
Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach
journal, April 2015
- Ramakrishnan, Raghunathan; Dral, Pavlo O.; Rupp, Matthias
- Journal of Chemical Theory and Computation, Vol. 11, Issue 5
ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB
journal, July 2015
- Maier, James A.; Martinez, Carmenza; Kasavajhala, Koushik
- Journal of Chemical Theory and Computation, Vol. 11, Issue 8
The atomic simulation environment—a Python library for working with atoms
journal, June 2017
- Hjorth Larsen, Ask; Jørgen Mortensen, Jens; Blomqvist, Jakob
- Journal of Physics: Condensed Matter, Vol. 29, Issue 27
Energy-free machine learning force field for aluminum
journal, August 2017
- Kruglov, Ivan; Sergeev, Oleg; Yanilkin, Alexey
- Scientific Reports, Vol. 7, Issue 1
The TensorMol-0.1 model chemistry: a neural network augmented with long-range physics
journal, January 2018
- Yao, Kun; Herr, John E.; Toth, David W.
- Chemical Science, Vol. 9, Issue 8
Ab Initio Investigation of O–H Dissociation from the Al–OH 2 Complex Using Molecular Dynamics and Neural Network Fitting
journal, January 2016
- Ho, Thi H.; Pham-Tran, Nguyen-Nguyen; Kawazoe, Yoshiyuki
- The Journal of Physical Chemistry A, Vol. 120, Issue 3
Metadynamics for training neural network model chemistries: A competitive assessment
journal, June 2018
- Herr, John E.; Yao, Kun; McIntyre, Ryker
- The Journal of Chemical Physics, Vol. 148, Issue 24
Calculation of properties with the coupled-cluster method
journal, January 1977
- Monkhorst, Hendrik J.
- International Journal of Quantum Chemistry, Vol. 12, Issue S11
Digitization of multistep organic synthesis in reactionware for on-demand pharmaceuticals
journal, January 2018
- Kitson, Philip J.; Marie, Guillaume; Francoia, Jean-Patrick
- Science, Vol. 359, Issue 6373
ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost
journal, January 2017
- Smith, J. S.; Isayev, O.; Roitberg, A. E.
- Chemical Science, Vol. 8, Issue 4
The Automation of Science
journal, April 2009
- King, Ross D.; Rowland, Jem; Oliver, Stephen G.
- Science, Vol. 324, Issue 5923
Structure of aqueous NaOH solutions: insights from neural-network-based molecular dynamics simulations
journal, January 2017
- Hellström, Matti; Behler, Jörg
- Physical Chemistry Chemical Physics, Vol. 19, Issue 1
Quantum-chemical insights from deep tensor neural networks
journal, January 2017
- Schütt, Kristof T.; Arbabzadah, Farhad; Chmiela, Stefan
- Nature Communications, Vol. 8, Issue 1
MyChEMBL: A Virtual Platform for Distributing Cheminformatics Tools and Open Data
journal, September 2014
- Davies, Mark; Nowotka, Michał; Papadatos, George
- Challenges, Vol. 5, Issue 2
Genetic Optimization of Training Sets for Improved Machine Learning Models of Molecular Properties
journal, March 2017
- Browning, Nicholas J.; Ramakrishnan, Raghunathan; von Lilienfeld, O. Anatole
- The Journal of Physical Chemistry Letters, Vol. 8, Issue 7
Pressure-induced phase transitions in silicon studied by neural network-based metadynamics simulations
journal, December 2008
- Behler, Jörg; Martoňák, Roman; Donadio, Davide
- physica status solidi (b), Vol. 245, Issue 12
Machine Learning Force Fields: Construction, Validation, and Outlook
journal, December 2016
- Botu, V.; Batra, R.; Chapman, J.
- The Journal of Physical Chemistry C, Vol. 121, Issue 1
Machine learning molecular dynamics for the simulation of infrared spectra
journal, January 2017
- Gastegger, Michael; Behler, Jörg; Marquetand, Philipp
- Chemical Science, Vol. 8, Issue 10
DrugBank 4.0: shedding new light on drug metabolism
journal, November 2013
- Law, Vivian; Knox, Craig; Djoumbou, Yannick
- Nucleic Acids Research, Vol. 42, Issue D1
Machine-learning approaches in drug discovery: methods and applications
journal, March 2015
- Lavecchia, Antonio
- Drug Discovery Today, Vol. 20, Issue 3
Machine-learning-assisted materials discovery using failed experiments
journal, May 2016
- Raccuglia, Paul; Elbert, Katherine C.; Adler, Philip D. F.
- Nature, Vol. 533, Issue 7601
Universal fragment descriptors for predicting properties of inorganic crystals
journal, June 2017
- Isayev, Olexandr; Oses, Corey; Toher, Cormac
- Nature Communications, Vol. 8, Issue 1
Quantum chemistry structures and properties of 134 kilo molecules
journal, August 2014
- Ramakrishnan, Raghunathan; Dral, Pavlo O.; Rupp, Matthias
- Scientific Data, Vol. 1, Issue 1
Active learning of linearly parametrized interatomic potentials
journal, December 2017
- Podryabinkin, Evgeny V.; Shapeev, Alexander V.
- Computational Materials Science, Vol. 140
Works referencing / citing this record:
Making machine learning a useful tool in the accelerated discovery of transition metal complexes
journal, July 2019
- Kulik, Heather J.
- WIREs Computational Molecular Science, Vol. 10, Issue 1
Machine learning and artificial neural network accelerated computational discoveries in materials science
journal, November 2019
- Hong, Yang; Hou, Bo; Jiang, Hengle
- WIREs Computational Molecular Science, Vol. 10, Issue 3
Active learning in materials science with emphasis on adaptive sampling using uncertainties for targeted design
journal, February 2019
- Lookman, Turab; Balachandran, Prasanna V.; Xue, Dezhen
- npj Computational Materials, Vol. 5, Issue 1
Machine learning enables long time scale molecular photodynamics simulations
journal, January 2019
- Westermayr, Julia; Gastegger, Michael; Menger, Maximilian F. S. J.
- Chemical Science, Vol. 10, Issue 35
IMPRESSION – prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy
journal, January 2020
- Gerrard, Will; Bratholm, Lars A.; Packer, Martin J.
- Chemical Science, Vol. 11, Issue 2
Guest Editorial: Special Topic on Data-Enabled Theoretical Chemistry
journal, June 2018
- Rupp, Matthias; von Lilienfeld, O. Anatole; Burke, Kieron
- The Journal of Chemical Physics, Vol. 148, Issue 24
Compressing physics with an autoencoder: Creating an atomic species representation to improve machine learning models in the chemical sciences
journal, August 2019
- Herr, John E.; Koh, Kevin; Yao, Kun
- The Journal of Chemical Physics, Vol. 151, Issue 8
Ring polymer molecular dynamics and active learning of moment tensor potential for gas-phase barrierless reactions: Application to S + H 2
journal, December 2019
- Novikov, Ivan S.; Shapeev, Alexander V.; Suleimanov, Yury V.
- The Journal of Chemical Physics, Vol. 151, Issue 22
From DFT to machine learning: recent approaches to materials science–a review
journal, May 2019
- Schleder, Gabriel R.; Padilha, Antonio C. M.; Acosta, Carlos Mera
- Journal of Physics: Materials, Vol. 2, Issue 3
Accessing thermal conductivity of complex compounds by machine learning interatomic potentials
journal, October 2019
- Korotaev, Pavel; Novoselov, Ivan; Yanilkin, Aleksey
- Physical Review B, Vol. 100, Issue 14
Constructing convex energy landscapes for atomistic structure optimization
journal, December 2019
- Chiriki, Siva; Christiansen, Mads-Peter V.; Hammer, B.
- Physical Review B, Vol. 100, Issue 23
Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network
journal, August 2019
- Zubatyuk, Roman; Smith, Justin S.; Leszczynski, Jerzy
- Science Advances, Vol. 5, Issue 8
Active Learning of Uniformly Accurate Inter-atomic Potentials for Materials Simulation
text, January 2018
- Zhang, Linfeng; Lin, De-Ye; Wang, Han
- arXiv
Machine learning enables long time scale molecular photodynamics simulations
text, January 2018
- Westermayr, Julia; Gastegger, Michael; Menger, Maximilian F. S. J.
- arXiv
IMPRESSION -- Prediction of NMR Parameters for 3-dimensional chemical structures using Machine Learning with near quantum chemical accuracy
preprint, January 2019
- Gerrard, Will; Bratholm, Lars Andersen; Packer, Martin
- arXiv
Ring Polymer Molecular Dynamics and Active Learning of Moment Tensor Potential for Gas-Phase Barrierless Reactions: Application to S + H2
text, January 2019
- Novikov, Ivan S.; Shapeev, Alexander V.; Suleimanov, Yury V.
- arXiv
Molecular Dynamics with Neural Network Potentials
book, January 2020
- Gastegger, Michael; Marquetand, Philipp
- Machine Learning Meets Quantum Physics
Liposome encapsulation circumvents the hepatic clearance mechanisms of all-trans-retinoic acid
journal, August 1994
- Mehta, Kapil; Sadeghi, Taraneh; McQueen, Teresa
- Leukemia Research, Vol. 18, Issue 8
Gaussian Process-Based Refinement of Dispersion Corrections
journal, October 2019
- Proppe, Jonny; Gugler, Stefan; Reiher, Markus
- Journal of Chemical Theory and Computation, Vol. 15, Issue 11
Machine learning enables long time scale molecular photodynamics simulations
journal, January 2019
- Westermayr, Julia; Gastegger, Michael; Menger, Maximilian F. S. J.
- Chemical Science, Vol. 10, Issue 35
Accurate and transferable multitask prediction of chemical properties with an atoms-in-molecules neural network
journal, August 2019
- Zubatyuk, Roman; Smith, Justin S.; Leszczynski, Jerzy
- Science Advances, Vol. 5, Issue 8
Deep Learning for Deep Chemistry: Optimizing the Prediction of Chemical Patterns
journal, November 2019
- Cova, Tânia F. G. G.; Pais, Alberto A. C. C.
- Frontiers in Chemistry, Vol. 7
Adversarial Active Learning for Deep Networks: a Margin Based Approach
preprint, January 2018
- Ducoffe, Melanie; Precioso, Frederic
- arXiv
Deep Ensemble Bayesian Active Learning : Addressing the Mode Collapse issue in Monte Carlo dropout via Ensembles
preprint, January 2018
- Pop, Remus; Fulop, Patric
- arXiv
Machine Learning of coarse-grained Molecular Dynamics Force Fields
preprint, January 2018
- Wang, Jiang; Olsson, Simon; Wehmeyer, Christoph
- arXiv
Molecular Dynamics with Neural-Network Potentials
preprint, January 2018
- Gastegger, Michael; Marquetand, Philipp
- arXiv
Machine Learning Prediction of DNA Charge Transport
text, January 2018
- Korol, Roman; Segal, Dvira
- arXiv
A Scalable Molecular Force Field Parameterization Method Based on Density Functional Theory and Quantum-Level Machine Learning
preprint, January 2019
- Galvelis, Raimondas; Doerr, Stefan; Damas, Joao M.
- arXiv
Incorporating electronic information into Machine Learning potential energy surfaces via approaching the ground-state electronic energy as a function of atom-based electronic populations
preprint, January 2020
- Xie, Xiaowei; Persson, Kristin A.; Small, David W.
- arXiv
Dropout Strikes Back: Improved Uncertainty Estimation via Diversity Sampling
preprint, January 2020
- Fedyanin, Kirill; Tsymbalov, Evgenii; Panov, Maxim
- arXiv
Machine Learning for Multi-fidelity Scale Bridging and Dynamical Simulations of Materials
preprint, January 2020
- Batra, Rohit; Sankaranarayanan, Subramanian
- arXiv
Opportunities and Challenges for Machine Learning in Materials Science
text, January 2020
- Morgan, Dane; Jacobs, Ryan
- arXiv
Deep Learning in Protein Structural Modeling and Design
preprint, January 2020
- Gao, Wenhao; Mahajan, Sai Pooja; Sulam, Jeremias
- arXiv
The MLIP package: Moment Tensor Potentials with MPI and Active Learning
preprint, January 2020
- Novikov, Ivan S.; Gubaev, Konstantin; Podryabinkin, Evgeny V.
- arXiv
Facilitating {\it ab initio} configurational sampling of multicomponent solids using an on-lattice neural network model and active learning
preprint, January 2020
- Kasamatsu, Shusuke; Motoyama, Yuichi; Yoshimi, Kazuyoshi
- arXiv
Training Data Set Refinement for the Machine Learning Potential of Li-Si Alloys via Structural Similarity Analysis
preprint, January 2021
- Xu, Nan; Li, Chen; Fang, Mandi
- arXiv
Figures / Tables found in this record: