
A Physical Model for Understanding the Activation of MoS2 Basal-plane Sulfur Atoms for the Hydrogen Evolution Reaction
Abstract
All the DFT calculations are done with the Vienna Ab Initio Simulation Package (VASP) using the projector augmented wave method. The Bayesian error estimation exchange-correlation functionals (BEEF) with van der Waals interactions are employed. A plane-wave cutoff energy of 400 eV is used together with PAW-PBE potentials where semi core p states are treated as valence. All the calculations allow for spin-polarization. The structures are relaxed until the force is converged to < 0.01 eV/Å. The lattice parameter of MoS2 unit cell, optimized with this functional, is 3.19 Å. A (4×4) supercell is used to model all the transition metal doped MoS2 systems studied here, including those with S vacancies. For calculations in the initial dopant structure exploration, the Brillouin zone is sampled with a 3x3x1 Monkhorst-Pack k-point mesh. A 6×6×1 Monkhorst-Pack k-point mesh is used for the H binding energy and density of states calculations. In all calculations, the vacuum layer is set as 15 Å to eliminate periodic interaction perpendicular to the basal plane.
- Authors:
-
 [1];
[1];
- Brookhaven National Lab. (BNL), Upton, NY (United States). Center for Functional Nanomaterials (CFN)
- Publication Date:
- Research Org.:
- Brookhaven National Lab. (BNL), Upton, NY (United States)
- Sponsoring Org.:
- USDOE Office of Science (SC), Basic Energy Sciences (BES)
- OSTI Identifier:
- 1631928
- Report Number(s):
- BNL-215970-2020-JAAM
Journal ID: ISSN 1089-5639
- Grant/Contract Number:
- SC0012704
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Journal of Physical Chemistry. A, Molecules, Spectroscopy, Kinetics, Environment, and General Theory
- Additional Journal Information:
- Journal Volume: 124; Journal Issue: 4; Journal ID: ISSN 1089-5639
- Publisher:
- American Chemical Society
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 77 NANOSCIENCE AND NANOTECHNOLOGY
Citation Formats
Liu, Mingjie, Wu, Qin, and Hybertsen, M. S. A Physical Model for Understanding the Activation of MoS2 Basal-plane Sulfur Atoms for the Hydrogen Evolution Reaction. United States: N. p., 2020.
Web. doi:10.26434/chemrxiv.11916168.
Liu, Mingjie, Wu, Qin, & Hybertsen, M. S. A Physical Model for Understanding the Activation of MoS2 Basal-plane Sulfur Atoms for the Hydrogen Evolution Reaction. United States. https://doi.org/10.26434/chemrxiv.11916168
Liu, Mingjie, Wu, Qin, and Hybertsen, M. S. Mon .
"A Physical Model for Understanding the Activation of MoS2 Basal-plane Sulfur Atoms for the Hydrogen Evolution Reaction". United States. https://doi.org/10.26434/chemrxiv.11916168. https://www.osti.gov/servlets/purl/1631928.
@article{osti_1631928,
title = {A Physical Model for Understanding the Activation of MoS2 Basal-plane Sulfur Atoms for the Hydrogen Evolution Reaction},
author = {Liu, Mingjie and Wu, Qin and Hybertsen, M. S.},
abstractNote = {All the DFT calculations are done with the Vienna Ab Initio Simulation Package (VASP) using the projector augmented wave method. The Bayesian error estimation exchange-correlation functionals (BEEF) with van der Waals interactions are employed. A plane-wave cutoff energy of 400 eV is used together with PAW-PBE potentials where semi core p states are treated as valence. All the calculations allow for spin-polarization. The structures are relaxed until the force is converged to < 0.01 eV/Å. The lattice parameter of MoS2 unit cell, optimized with this functional, is 3.19 Å. A (4×4) supercell is used to model all the transition metal doped MoS2 systems studied here, including those with S vacancies. For calculations in the initial dopant structure exploration, the Brillouin zone is sampled with a 3x3x1 Monkhorst-Pack k-point mesh. A 6×6×1 Monkhorst-Pack k-point mesh is used for the H binding energy and density of states calculations. In all calculations, the vacuum layer is set as 15 Å to eliminate periodic interaction perpendicular to the basal plane.},
doi = {10.26434/chemrxiv.11916168},
journal = {Journal of Physical Chemistry. A, Molecules, Spectroscopy, Kinetics, Environment, and General Theory},
number = 4,
volume = 124,
place = {United States},
year = {Mon Feb 03 00:00:00 EST 2020},
month = {Mon Feb 03 00:00:00 EST 2020}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalFigures / Tables:
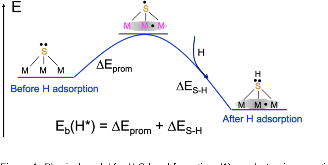 Figure 1: Physical model for H-S bond formation: (1) an electronic promotion process that pushes an electron from the full valence shell of S to neighboring metal atoms; (2) H binding to fill the partially open valence shell of the S site.
Figure 1: Physical model for H-S bond formation: (1) an electronic promotion process that pushes an electron from the full valence shell of S to neighboring metal atoms; (2) H binding to fill the partially open valence shell of the S site.
Figures / Tables found in this record: