
Structure and electronic properties of rare earth DOBDC metal–organic-frameworks
Abstract
In our report, we apply density functional theory (DFT) to investigate rare-earth metal organic frameworks (RE-MOFs), RE12(μ3-OH)16(C8O6H4)8(C8O6H5)4 (RE = Y, Eu, Tb, Yb), and characterize the level of theory needed to accurately predict structural and electronic properties in MOF materials with 4f-electrons. A two-step calculation approach of geometry optimization with spin-restricted DFT and large core potential (LCPs), and detailed electronic structures with spin-unrestricted DFT with a full valence potential + Hubbard U correction is investigated. Spin-restricted DFT with LCPs resulted in good agreement between experimental lattice parameters and optimized geometries, while a full valence potential is necessary for accurate representation of the electronic structure. The electronic structure of Eu-DOBDC MOF suggested a strong dependence on the treatment of highly localized 4f-electrons and spin polarization, as well as variation within a range of Hubbard corrections (U = 1–9 eV). For Hubbard corrected spin-unrestricted calculations, a U value of 1–4 eV maintains the non-metallic character of the band gap with slight deviations in f-orbital energetics. When compared with experimentally reported results, the importance of the full valence calculation and the Hubbard correction in correctly predicting the electronic structure is highlighted.
- Publication Date:
- Research Org.:
- Energy Frontier Research Centers (EFRC) (United States). Center for Understanding and Control of Acid Gas-induced Evolution of Materials for Energy (UNCAGE-ME); Sandia National Lab. (SNL-NM), Albuquerque, NM (United States)
- Sponsoring Org.:
- USDOE Office of Science (SC), Basic Energy Sciences (BES); USDOE National Nuclear Security Administration (NNSA)
- OSTI Identifier:
- 1574447
- Alternate Identifier(s):
- OSTI ID: 1570052
- Report Number(s):
- SAND-2019-12496J
Journal ID: ISSN 1463-9076; PPCPFQ; 680399; TRN: US2100017
- Grant/Contract Number:
- AC04-94AL85000; SC0012577; NA0003525; UNCAGE-ME Energy Frontier Research Center (EFRC) A; #DE-SC0012577
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Physical Chemistry Chemical Physics. PCCP
- Additional Journal Information:
- Journal Volume: 21; Journal Issue: 41; Journal ID: ISSN 1463-9076
- Publisher:
- Royal Society of Chemistry
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 37 INORGANIC, ORGANIC, PHYSICAL, AND ANALYTICAL CHEMISTRY
Citation Formats
Vogel, Dayton J., Sava Gallis, Dorina F., Nenoff, Tina M., and Rimsza, Jessica M. Structure and electronic properties of rare earth DOBDC metal–organic-frameworks. United States: N. p., 2019.
Web. doi:10.1039/C9CP04038B.
Vogel, Dayton J., Sava Gallis, Dorina F., Nenoff, Tina M., & Rimsza, Jessica M. Structure and electronic properties of rare earth DOBDC metal–organic-frameworks. United States. https://doi.org/10.1039/C9CP04038B
Vogel, Dayton J., Sava Gallis, Dorina F., Nenoff, Tina M., and Rimsza, Jessica M. Fri .
"Structure and electronic properties of rare earth DOBDC metal–organic-frameworks". United States. https://doi.org/10.1039/C9CP04038B. https://www.osti.gov/servlets/purl/1574447.
@article{osti_1574447,
title = {Structure and electronic properties of rare earth DOBDC metal–organic-frameworks},
author = {Vogel, Dayton J. and Sava Gallis, Dorina F. and Nenoff, Tina M. and Rimsza, Jessica M.},
abstractNote = {In our report, we apply density functional theory (DFT) to investigate rare-earth metal organic frameworks (RE-MOFs), RE12(μ3-OH)16(C8O6H4)8(C8O6H5)4 (RE = Y, Eu, Tb, Yb), and characterize the level of theory needed to accurately predict structural and electronic properties in MOF materials with 4f-electrons. A two-step calculation approach of geometry optimization with spin-restricted DFT and large core potential (LCPs), and detailed electronic structures with spin-unrestricted DFT with a full valence potential + Hubbard U correction is investigated. Spin-restricted DFT with LCPs resulted in good agreement between experimental lattice parameters and optimized geometries, while a full valence potential is necessary for accurate representation of the electronic structure. The electronic structure of Eu-DOBDC MOF suggested a strong dependence on the treatment of highly localized 4f-electrons and spin polarization, as well as variation within a range of Hubbard corrections (U = 1–9 eV). For Hubbard corrected spin-unrestricted calculations, a U value of 1–4 eV maintains the non-metallic character of the band gap with slight deviations in f-orbital energetics. When compared with experimentally reported results, the importance of the full valence calculation and the Hubbard correction in correctly predicting the electronic structure is highlighted.},
doi = {10.1039/C9CP04038B},
journal = {Physical Chemistry Chemical Physics. PCCP},
number = 41,
volume = 21,
place = {United States},
year = {Fri Oct 11 00:00:00 EDT 2019},
month = {Fri Oct 11 00:00:00 EDT 2019}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
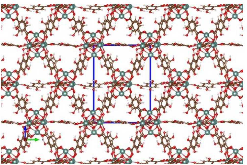 Figure 1: Periodic 3D structure of activated Eu-DOBDC with a single unit cell centrally located and highlighted by a blue box. Colors: Eu(green), 0(red), C(brown), and H(white)
Figure 1: Periodic 3D structure of activated Eu-DOBDC with a single unit cell centrally located and highlighted by a blue box. Colors: Eu(green), 0(red), C(brown), and H(white)
Works referenced in this record:
Flexibility in metal-organic framework materials: Impact on sorption properties
journal, August 2005
- Fletcher, Ashleigh J.; Thomas, K. Mark; Rosseinsky, Matthew J.
- Journal of Solid State Chemistry, Vol. 178, Issue 8
Projector augmented-wave method
journal, December 1994
- Blöchl, P. E.
- Physical Review B, Vol. 50, Issue 24, p. 17953-17979
Acid Gas Stability of Zeolitic Imidazolate Frameworks: Generalized Kinetic and Thermodynamic Characteristics
journal, May 2018
- Bhattacharyya, Souryadeep; Han, Rebecca; Kim, Wun-Gwi
- Chemistry of Materials, Vol. 30, Issue 12
Metal–organic frameworks: functional luminescent and photonic materials for sensing applications
journal, January 2017
- Lustig, William P.; Mukherjee, Soumya; Rudd, Nathan D.
- Chemical Society Reviews, Vol. 46, Issue 11
Assessing Hubbard-corrected AM05+U and PBEsol+U density functionals for strongly correlated oxides CeO 2 and Ce 2 O 3
journal, January 2016
- Weck, Philippe F.; Kim, Eunja
- Physical Chemistry Chemical Physics, Vol. 18, Issue 38
From ultrasoft pseudopotentials to the projector augmented-wave method
journal, January 1999
- Kresse, G.; Joubert, D.
- Physical Review B, Vol. 59, Issue 3, p. 1758-1775
Small-Molecule Adsorption in Open-Site Metal–Organic Frameworks: A Systematic Density Functional Theory Study for Rational Design
journal, January 2015
- Lee, Kyuho; Howe, Joshua D.; Lin, Li-Chiang
- Chemistry of Materials, Vol. 27, Issue 3, p. 668-678
Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study
journal, January 1998
- Dudarev, S. L.; Botton, G. A.; Savrasov, S. Y.
- Physical Review B, Vol. 57, Issue 3, p. 1505-1509
The magnetic moments and electronic spectra of lanthanide chelates of 2-thenoyltrifluoroacetone
journal, October 1983
- Hamer, Andrew M.; Livingstone, Stanley E.
- Transition Metal Chemistry, Vol. 8, Issue 5
Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators
journal, August 1995
- Liechtenstein, A. I.; Anisimov, V. I.; Zaanen, J.
- Physical Review B, Vol. 52, Issue 8
NMR Spectroscopy Reveals Adsorbate Binding Sites in the Metal–Organic Framework UiO-66(Zr)
journal, March 2018
- Nandy, Aditya; Forse, Alexander C.; Witherspoon, Velencia J.
- The Journal of Physical Chemistry C, Vol. 122, Issue 15
Recent advances in gas storage and separation using metal–organic frameworks
journal, March 2018
- Li, Hao; Wang, Kecheng; Sun, Yujia
- Materials Today, Vol. 21, Issue 2
Biocompatible MOFs with high absolute quantum yield for bioimaging in the second near infrared window
journal, January 2018
- Sava Gallis, Dorina F.; Butler, Kimberly S.; Rohwer, Lauren E. S.
- CrystEngComm, Vol. 20, Issue 39
Thermoelectric properties for AA- and AB-stacking of a carbon nitride polymorph (C 3 N 4 )
journal, January 2014
- Reshak, A. H.
- RSC Adv., Vol. 4, Issue 108
Photoelectrical properties and the electronic structure of Tl1−xIn1−xSnxSe2 (x = 0, 0.1, 0.2, 0.25) single crystalline alloys
journal, January 2013
- Davydyuk, G. E.; Khyzhun, O. Y.; Reshak, A. H.
- Physical Chemistry Chemical Physics, Vol. 15, Issue 18
DFT Calculation of Russell–Saunders Splitting for Lanthanide Ions Doped in Hexagonal (β)-NaYF 4 Nanocrystals
journal, August 2013
- Yao, Ge; Berry, Mary T.; May, P. Stanley
- The Journal of Physical Chemistry C, Vol. 117, Issue 33
Efficient MOF-based degradation of organophosphorus compounds in non-aqueous environments
journal, January 2018
- Sava Gallis, Dorina F.; Harvey, Jacob A.; Pearce, Charles J.
- Journal of Materials Chemistry A, Vol. 6, Issue 7
Ab initiomolecular dynamics for liquid metals
journal, January 1993
- Kresse, G.; Hafner, J.
- Physical Review B, Vol. 47, Issue 1, p. 558-561
Density-functional theory and NiO photoemission spectra
journal, December 1993
- Anisimov, V. I.; Solovyev, I. V.; Korotin, M. A.
- Physical Review B, Vol. 48, Issue 23
On The Density Functional Theory Treatment of Lanthanide Coordination Compounds: A Comparative Study in a Series of Cu–Ln (Ln = Gd, Tb, Lu) Binuclear Complexes
journal, July 2017
- Ferbinteanu, Marilena; Stroppa, Alessandro; Scarrozza, Marco
- Inorganic Chemistry, Vol. 56, Issue 16
Interpretation of europium(III) spectra
journal, July 2015
- Binnemans, Koen
- Coordination Chemistry Reviews, Vol. 295
Efficacy of Density Functionals and Relativistic Effective Core Potentials for Lanthanide-Containing Species: The Ln54 Molecule Set
journal, May 2017
- Aebersold, Lucas E.; Yuwono, Stephen H.; Schoendorff, George
- Journal of Chemical Theory and Computation, Vol. 13, Issue 6
Assessment of Density Functionals for Computing Thermodynamic Properties of Lanthanide Complexes
journal, August 2017
- Jaoul, Arnaud; Nocton, Grégory; Clavaguéra, Carine
- ChemPhysChem, Vol. 18, Issue 19
Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research
journal, January 2019
- Yang, Dong; Gates, Bruce C.
- ACS Catalysis, Vol. 9, Issue 3
A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu
journal, April 2010
- Grimme, Stefan; Antony, Jens; Ehrlich, Stephan
- The Journal of Chemical Physics, Vol. 132, Issue 15
Physical Content of the Exact Kohn-Sham Orbital Energies: Band Gaps and Derivative Discontinuities
journal, November 1983
- Perdew, John P.; Levy, Mel
- Physical Review Letters, Vol. 51, Issue 20
Effect of the damping function in dispersion corrected density functional theory
journal, March 2011
- Grimme, Stefan; Ehrlich, Stephan; Goerigk, Lars
- Journal of Computational Chemistry, Vol. 32, Issue 7
Performance of DFT+ U method for prediction of structural and thermodynamic parameters of monazite-type ceramics
journal, April 2014
- Blanca Romero, Ariadna; Kowalski, Piotr M.; Beridze, George
- Journal of Computational Chemistry, Vol. 35, Issue 18
Effect of Metal in M 3 (btc) 2 and M 2 (dobdc) MOFs for O 2 /N 2 Separations: A Combined Density Functional Theory and Experimental Study
journal, March 2015
- Parkes, Marie V.; Sava Gallis, Dorina F.; Greathouse, Jeffery A.
- The Journal of Physical Chemistry C, Vol. 119, Issue 12
Dispersion of linear and nonlinear optical susceptibilities and the hyperpolarizability of 3-methyl-4-phenyl-5-(2-pyridyl)-1,2,4-triazole
journal, January 2011
- Reshak, Ali. H.; Stys, D.; Auluck, S.
- Phys. Chem. Chem. Phys., Vol. 13, Issue 7
Computational Screening of Metal–Organic Frameworks for Membrane-Based CO 2 /N 2 /H 2 O Separations: Best Materials for Flue Gas Separation
journal, March 2018
- Daglar, Hilal; Keskin, Seda
- The Journal of Physical Chemistry C, Vol. 122, Issue 30
Zr-based metal–organic frameworks: design, synthesis, structure, and applications
journal, January 2016
- Bai, Yan; Dou, Yibo; Xie, Lin-Hua
- Chemical Society Reviews, Vol. 45, Issue 8
First-principles Hubbard U approach for small molecule binding in metal-organic frameworks
journal, May 2016
- Mann, Gregory W.; Lee, Kyuho; Cococcioni, Matteo
- The Journal of Chemical Physics, Vol. 144, Issue 17
Multifunctional, Tunable Metal–Organic Framework Materials Platform for Bioimaging Applications
journal, June 2017
- Sava Gallis, Dorina F.; Rohwer, Lauren E. S.; Rodriguez, Mark A.
- ACS Applied Materials & Interfaces, Vol. 9, Issue 27
Insights into the Stability of Zeolitic Imidazolate Frameworks in Humid Acidic Environments from First-Principles Calculations
journal, February 2018
- Han, Chu; Zhang, Chenyang; Tymińska, Nina
- The Journal of Physical Chemistry C, Vol. 122, Issue 8
Electronic structure and magnetic properties of lanthanide 3+ cations
journal, June 2013
- Kurzen, Helena; Bovigny, Laura; Bulloni, Claudio
- Chemical Physics Letters, Vol. 574
Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set
journal, July 1996
- Kresse, G.; Furthmüller, J.
- Computational Materials Science, Vol. 6, Issue 1, p. 15-50
SO x /NO x Removal from Flue Gas Streams by Solid Adsorbents: A Review of Current Challenges and Future Directions
journal, August 2015
- Rezaei, Fateme; Rownaghi, Ali A.; Monjezi, Saman
- Energy & Fuels, Vol. 29, Issue 9
Lanthanide-Functionalized Metal–Organic Framework Hybrid Systems To Create Multiple Luminescent Centers for Chemical Sensing
journal, October 2017
- Yan, Bing
- Accounts of Chemical Research, Vol. 50, Issue 11
Stability of Zeolitic Imidazolate Frameworks in NO 2
journal, January 2019
- Bhattacharyya, Souryadeep; Han, Rebecca; Joshi, Jayraj N.
- The Journal of Physical Chemistry C, Vol. 123, Issue 4
Revised Ionic Radii of Lanthanoid(III) Ions in Aqueous Solution
journal, May 2011
- D’Angelo, Paola; Zitolo, Andrea; Migliorati, Valentina
- Inorganic Chemistry, Vol. 50, Issue 10
Ionization Energies of Lanthanides
journal, August 2010
- Lang, Peter F.; Smith, Barry C.
- Journal of Chemical Education, Vol. 87, Issue 8
Ab initio study of TaON, an active photocatalyst under visible light irradiation
journal, January 2014
- Reshak, A. H.
- Physical Chemistry Chemical Physics, Vol. 16, Issue 22
Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
journal, October 1996
- Kresse, G.; Furthmüller, J.
- Physical Review B, Vol. 54, Issue 16, p. 11169-11186
Towards metal–organic framework based field effect chemical sensors: UiO-66-NH 2 for nerve agent detection
journal, January 2016
- Stassen, I.; Bueken, B.; Reinsch, H.
- Chemical Science, Vol. 7, Issue 9
Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium
journal, May 1994
- Kresse, G.; Hafner, J.
- Physical Review B, Vol. 49, Issue 20, p. 14251-14269
Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces
journal, April 2008
- Perdew, John P.; Ruzsinszky, Adrienn; Csonka, Gábor I.
- Physical Review Letters, Vol. 100, Issue 13
Metal–Organic Frameworks for Separations
journal, September 2011
- Li, Jian-Rong; Sculley, Julian; Zhou, Hong-Cai
- Chemical Reviews, Vol. 112, Issue 2, p. 869-932
Two Lanthanide Metal–Organic Frameworks as Remarkably Selective and Sensitive Bifunctional Luminescence Sensor for Metal Ions and Small Organic Molecules
journal, January 2017
- Yan, Wei; Zhang, Chuanlei; Chen, Shuguang
- ACS Applied Materials & Interfaces, Vol. 9, Issue 2
In Situ Probes of Capture and Decomposition of Chemical Warfare Agent Simulants by Zr-Based Metal Organic Frameworks
journal, January 2017
- Plonka, Anna M.; Wang, Qi; Gordon, Wesley O.
- Journal of the American Chemical Society, Vol. 139, Issue 2
Ab initio calculation of energy levels of trivalent lanthanide ions
journal, January 2018
- Freidzon, Alexandra Ya.; Kurbatov, Ilia A.; Vovna, Vitaliy I.
- Physical Chemistry Chemical Physics, Vol. 20, Issue 21
Theoretical design of highly luminescent europium (III) complexes: A factorial study
journal, January 2011
- Dutra, José Diogo L.; Gimenez, Iara F.; Junior, Nivan B. da Costa
- Journal of Photochemistry and Photobiology A: Chemistry, Vol. 217, Issue 2-3
Density functional calculations of lanthanide oxides
journal, June 1995
- Wang, S. G.; Pan, D. K.; Schwarz, W. H. E.
- The Journal of Chemical Physics, Vol. 102, Issue 23
The Chemistry and Applications of Metal-Organic Frameworks
journal, August 2013
- Furukawa, H.; Cordova, K. E.; O'Keeffe, M.
- Science, Vol. 341, Issue 6149, p. 1230444-1230444
Flue gas treatment via CO2 adsorption
journal, July 2011
- Sayari, Abdelhamid; Belmabkhout, Youssef; Serna-Guerrero, Rodrigo
- Chemical Engineering Journal, Vol. 171, Issue 3
Mechanical properties of metal dihydrides
journal, February 2016
- Schultz, Peter A.; Snow, Clark S.
- Modelling and Simulation in Materials Science and Engineering, Vol. 24, Issue 3
Defect and Linker Effects on the Binding of Organophosphorous Compounds in UiO-66 and Rare-Earth MOFs
journal, November 2018
- Harvey, Jacob A.; Greathouse, Jeffery A.; Sava Gallis, Dorina F.
- The Journal of Physical Chemistry C, Vol. 122, Issue 47
Ab initio molecular dynamics determination of competitive O 2 vs. N 2 adsorption at open metal sites of M 2 (dobdc)
journal, January 2016
- Parkes, Marie V.; Greathouse, Jeffery A.; Hart, David B.
- Physical Chemistry Chemical Physics, Vol. 18, Issue 16
Luminescence properties and DFT calculations of lanthanide(III) complexes (Ln = La, Nd, Sm, Eu, Gd, Tb, Dy) with 2,6-bis(5-methyl-benzimidazol-2-yl)pyridine
journal, July 2018
- Cruz-Navarro, Antonio; Rivera, José María; Durán-Hernández, Jesús
- Journal of Molecular Structure, Vol. 1164
Zeolitic Imidazolate Frameworks: Next-Generation Materials for Energy-Efficient Gas Separations
journal, October 2014
- Pimentel, Brian R.; Parulkar, Aamena; Zhou, Er-kang
- ChemSusChem, Vol. 7, Issue 12
Direct solid-phase synthesis of molecular heterooligonuclear lanthanoid-complexes
journal, March 2020
- Kreidt, Elisabeth; Leis, Wolfgang; Seitz, Michael
- Nature Communications, Vol. 11, Issue 1
Voltage- and time-dependent valence state transition in cobalt oxide catalysts during the oxygen evolution reaction
journal, April 2020
- Zhou, Jing; Zhang, Linjuan; Huang, Yu-Cheng
- Nature Communications, Vol. 11, Issue 1
Reducing Dzyaloshinskii-Moriya interaction and field-free spin-orbit torque switching in synthetic antiferromagnets
journal, May 2021
- Chen, Ruyi; Cui, Qirui; Liao, Liyang
- Nature Communications, Vol. 12, Issue 1
High-resolution X-ray luminescence extension imaging
journal, February 2021
- Ou, Xiangyu; Qin, Xian; Huang, Bolong
- Nature, Vol. 590, Issue 7846
Electronic structure of AlFeN films exhibiting crystallographic orientation change from c- to a-axis with Fe concentrations and annealing effect
journal, February 2020
- Tatemizo, Nobuyuki; Imada, Saki; Okahara, Kizuna
- Scientific Reports, Vol. 10, Issue 1
Figures / Tables found in this record: