
Accurate and balanced anisotropic Gaussian type orbital basis sets for atoms in strong magnetic fields
Abstract
In high magnetic field calculations, anisotropic Gaussian type orbital (AGTO) basis functions are capable of reconciling the competing demands of the spherically symmetric Coulombic interaction and cylindrical magnetic (B field) confinement. However, the best available a priori procedure for composing highly accurate AGTO sets for atoms in a strong B field [W. Zhu et al., Phys. Rev. A 90, 022504 (2014)] yields very large basis sets. Their size is problematical for use in any calculation with unfavorable computational cost scaling. Here we provide an alternative constructive procedure. It is based upon analysis of the underlying physics of atoms in B fields that allow identification of several principles for the construction of AGTO basis sets. Aided by numerical optimization and parameter fitting, followed by fine tuning of fitting parameters, we devise formulae for generating accurate AGTO basis sets in an arbitrary B field. For the hydrogen iso-electronic sequence, a set depends on B field strength, nuclear charge, and orbital quantum numbers. For multi-electron systems, the basis set formulae also include adjustment to account for orbital occupations. Tests of the new basis sets for atoms H through C (1 ≤ Z ≤ 6) and ions Li+, Be+, and B+, in a widemore »
- Publication Date:
- Research Org.:
- Univ. of Florida, Gainesville, FL (United States)
- Sponsoring Org.:
- USDOE
- OSTI Identifier:
- 1512938
- Alternate Identifier(s):
- OSTI ID: 1414872
- Grant/Contract Number:
- SC0002139; SC- 0002139
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Journal of Chemical Physics
- Additional Journal Information:
- Journal Volume: 147; Journal Issue: 24; Journal ID: ISSN 0021-9606
- Publisher:
- American Institute of Physics (AIP)
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 71 CLASSICAL AND QUANTUM MECHANICS, GENERAL PHYSICS
Citation Formats
Zhu, Wuming, and Trickey, S. B. Accurate and balanced anisotropic Gaussian type orbital basis sets for atoms in strong magnetic fields. United States: N. p., 2017.
Web. doi:10.1063/1.5004713.
Zhu, Wuming, & Trickey, S. B. Accurate and balanced anisotropic Gaussian type orbital basis sets for atoms in strong magnetic fields. United States. https://doi.org/10.1063/1.5004713
Zhu, Wuming, and Trickey, S. B. Tue .
"Accurate and balanced anisotropic Gaussian type orbital basis sets for atoms in strong magnetic fields". United States. https://doi.org/10.1063/1.5004713. https://www.osti.gov/servlets/purl/1512938.
@article{osti_1512938,
title = {Accurate and balanced anisotropic Gaussian type orbital basis sets for atoms in strong magnetic fields},
author = {Zhu, Wuming and Trickey, S. B.},
abstractNote = {In high magnetic field calculations, anisotropic Gaussian type orbital (AGTO) basis functions are capable of reconciling the competing demands of the spherically symmetric Coulombic interaction and cylindrical magnetic (B field) confinement. However, the best available a priori procedure for composing highly accurate AGTO sets for atoms in a strong B field [W. Zhu et al., Phys. Rev. A 90, 022504 (2014)] yields very large basis sets. Their size is problematical for use in any calculation with unfavorable computational cost scaling. Here we provide an alternative constructive procedure. It is based upon analysis of the underlying physics of atoms in B fields that allow identification of several principles for the construction of AGTO basis sets. Aided by numerical optimization and parameter fitting, followed by fine tuning of fitting parameters, we devise formulae for generating accurate AGTO basis sets in an arbitrary B field. For the hydrogen iso-electronic sequence, a set depends on B field strength, nuclear charge, and orbital quantum numbers. For multi-electron systems, the basis set formulae also include adjustment to account for orbital occupations. Tests of the new basis sets for atoms H through C (1 ≤ Z ≤ 6) and ions Li+, Be+, and B+, in a wide B field range (0 ≤ B ≤ 2000 a.u.), show an accuracy better than a few μhartree for single-electron systems and a few hundredths to a few mHs for multi-electron atoms. The relative errors are similar for different atoms and ions in a large B field range, from a few to a couple of tens of millionths, thereby confirming rather uniform accuracy across the nuclear charge Z and B field strength values. Residual basis set errors are two to three orders of magnitude smaller than the electronic correlation energies in multi-electron atoms, a signal of the usefulness of the new AGTO basis sets in correlated wavefunction or density functional calculations for atomic and molecular systems in an external strong B field},
doi = {10.1063/1.5004713},
journal = {Journal of Chemical Physics},
number = 24,
volume = 147,
place = {United States},
year = {Tue Dec 26 00:00:00 EST 2017},
month = {Tue Dec 26 00:00:00 EST 2017}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
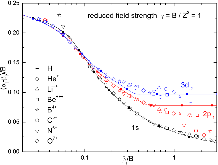 FIG. 1: Fitting the optimized exponents of AGTO basis functions for hydrogen atom and hydrogen-like ions in reduced magnetic field strength γ = 1. Empty symbols are optimized exponents from searching {αj} space. Solid curves are fits to those data points, generated by Eqs. (10)–(13). Filled squares are the AGTOmore »
FIG. 1: Fitting the optimized exponents of AGTO basis functions for hydrogen atom and hydrogen-like ions in reduced magnetic field strength γ = 1. Empty symbols are optimized exponents from searching {αj} space. Solid curves are fits to those data points, generated by Eqs. (10)–(13). Filled squares are the AGTOmore »
Works referenced in this record:
Hydrogen-Like Systems in Arbitrary Magnetic Fields A Variational Approch
journal, May 1979
- Aldrich, C.; Greene, R. L.
- Physica Status Solidi (b), Vol. 93, Issue 1
Full-core-plus-correlation method in cylindrical coordinates: Lithium atom in strong magnetic fields
journal, March 2007
- Wang, Xiaofeng; Qiao, Haoxue
- Physical Review A, Vol. 75, Issue 3
Ground states of H, He,…, Ne, and their singly positive ions in strong magnetic fields: The high-field regime
journal, January 2000
- Ivanov, M. V.; Schmelcher, P.
- Physical Review A, Vol. 61, Issue 2
Beryllium in strong magnetic fields
journal, August 2004
- Al-Hujaj, Omar-Alexander; Schmelcher, Peter
- Physical Review A, Vol. 70, Issue 2
B-spline algorithm for magnetized multielectron atomic structures
journal, March 2008
- Zhao, L. B.; Stancil, P. C.
- Physical Review A, Vol. 77, Issue 3
Exact solution for a hydrogen atom in a magnetic field of arbitrary strength
journal, July 1996
- Kravchenko, Yu. P.; Liberman, M. A.; Johansson, B.
- Physical Review A, Vol. 54, Issue 1
The beryllium atom and beryllium positive ion in strong magnetic fields
journal, June 2001
- Ivanov, M. V.; Schmelcher, P.
- The European Physical Journal D, Vol. 14, Issue 3
Self‐Consistent Molecular‐Orbital Methods. I. Use of Gaussian Expansions of Slater‐Type Atomic Orbitals
journal, September 1969
- Hehre, W. J.; Stewart, R. F.; Pople, J. A.
- The Journal of Chemical Physics, Vol. 51, Issue 6
The helium atom in a strong magnetic field
journal, January 1999
- Becken, W.; Schmelcher, P.; Diakonos, F. K.
- Journal of Physics B: Atomic, Molecular and Optical Physics, Vol. 32, Issue 6
Lithium in strong magnetic fields
journal, September 2004
- Al-Hujaj, Omar-Alexander; Schmelcher, Peter
- Physical Review A, Vol. 70, Issue 3
Gaussian basis sets for use in correlated molecular calculations. V. Core‐valence basis sets for boron through neon
journal, September 1995
- Woon, David E.; Dunning, Thom H.
- The Journal of Chemical Physics, Vol. 103, Issue 11
Density-functional calculations in a two-dimensional finite-element basis for atoms in very strong magnetic fields: Energy values
journal, February 2002
- Braun, M.
- Physical Review A, Vol. 65, Issue 3
Spectrum of neutral helium in strong magnetic fields
journal, April 1999
- Jones, Matthew D.; Ortiz, Gerardo; Ceperley, David M.
- Physical Review A, Vol. 59, Issue 4
Density-functional-theory calculations of matter in strong magnetic fields. I. Atoms and molecules
journal, December 2006
- Medin, Zach; Lai, Dong
- Physical Review A, Vol. 74, Issue 6
Relativistic and nonrelativistic finite-basis-set calculations of low-lying levels of hydrogenic atoms in intense magnetic fields
journal, February 1992
- Chen, Zonghua; Goldman, S. P.
- Physical Review A, Vol. 45, Issue 3
Coupled-cluster theory for atoms and molecules in strong magnetic fields
journal, August 2015
- Stopkowicz, Stella; Gauss, Jürgen; Lange, Kai K.
- The Journal of Chemical Physics, Vol. 143, Issue 7
Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen
journal, January 1989
- Dunning, Thom H.
- The Journal of Chemical Physics, Vol. 90, Issue 2
Effective convergence to complete orbital bases and to the atomic Hartree–Fock limit through systematic sequences of Gaussian primitives
journal, November 1979
- Schmidt, M. W.; Ruedenberg, K.
- The Journal of Chemical Physics, Vol. 71, Issue 10
Self‐Consistent Molecular‐Orbital Methods. IX. An Extended Gaussian‐Type Basis for Molecular‐Orbital Studies of Organic Molecules
journal, January 1971
- Ditchfield, R.; Hehre, W. J.; Pople, J. A.
- The Journal of Chemical Physics, Vol. 54, Issue 2
Accurate correlation consistent basis sets for molecular core–valence correlation effects: The second row atoms Al–Ar, and the first row atoms B–Ne revisited
journal, December 2002
- Peterson, Kirk A.; Dunning, Thom H.
- The Journal of Chemical Physics, Vol. 117, Issue 23
Hartree-Fock studies of atoms in strong magnetic fields
journal, July 1996
- Jones, Matthew D.; Ortiz, Gerardo; Ceperley, David M.
- Physical Review A, Vol. 54, Issue 1
Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions
journal, January 1980
- Krishnan, R.; Binkley, J. S.; Seeger, R.
- The Journal of Chemical Physics, Vol. 72, Issue 1
Molecules in strong magnetic fields: Properties of atomic orbitals
journal, February 1988
- Schmelcher, P.; Cederbaum, L. S.
- Physical Review A, Vol. 37, Issue 3
Ground state of the carbon atom in strong magnetic fields
journal, November 1999
- Ivanov, M. V.; Schmelcher, P.
- Physical Review A, Vol. 60, Issue 5
Application of Gaussian-type basis sets to ab initio calculations in strong magnetic fields
journal, January 1997
- Kravchenko, Yu. P.; Liberman, M. A.
- International Journal of Quantum Chemistry, Vol. 64, Issue 5
Hydrogen and helium atoms in strong magnetic fields
journal, January 2009
- Thirumalai, Anand; Heyl, Jeremy S.
- Physical Review A, Vol. 79, Issue 1
"Excitonic" matter in a superstrong magnetic field
journal, April 1974
- Chui, S. T.
- Physical Review B, Vol. 9, Issue 8
The boron atom and boron positive ion in strong magnetic fields
journal, May 2001
- Ivanov, M. V.; Schmelcher, P.
- Journal of Physics B: Atomic, Molecular and Optical Physics, Vol. 34, Issue 10
Non-zero angular momentum states of the helium atom in a strong magnetic field
journal, January 2000
- Becken, W.; Schmelcher, P.
- Journal of Physics B: Atomic, Molecular and Optical Physics, Vol. 33, Issue 3
Atomic orbital basis set optimization for ab initio calculations of molecules with hydrogen atoms in strong magnetic fields
journal, February 1994
- Kappes, U.; Schmelcher, P.
- The Journal of Chemical Physics, Vol. 100, Issue 4
Ground state of the lithium atom in strong magnetic fields
journal, May 1998
- Ivanov, M. V.; Schmelcher, P.
- Physical Review A, Vol. 57, Issue 5
Theory of the Quadratic Zeeman Effect
journal, January 1939
- Schiff, L. I.; Snyder, H.
- Physical Review, Vol. 55, Issue 1
Energy Spectrum of Hydrogen-Like Atoms in a Strong Magnetic Field
journal, June 1974
- Surmelian, G. L.; O'Conner, R. F.
- The Astrophysical Journal, Vol. 190
Multichannel density-functional calculations for atoms and atomic chains in magnetic fields of compact stars
journal, June 1996
- Relovsky, B. M.; Ruder, H.
- Physical Review A, Vol. 53, Issue 6
Higher-angular-momentum states of the helium atom in a strong magnetic field
journal, April 2001
- Becken, W.; Schmelcher, P.
- Physical Review A, Vol. 63, Issue 5
Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules
journal, March 1972
- Hehre, W. J.; Ditchfield, R.; Pople, J. A.
- The Journal of Chemical Physics, Vol. 56, Issue 5, p. 2257-2261
Works referencing / citing this record:
Kohn–Sham energy decomposition for molecules in a magnetic field
journal, May 2018
- Reimann, Sarah; Borgoo, Alex; Austad, Jon
- Molecular Physics, Vol. 117, Issue 1
Fully numerical electronic structure calculations on diatomic molecules in weak to strong magnetic fields
journal, March 2019
- Lehtola, Susi; Dimitrova, Maria; Sundholm, Dage
- Molecular Physics, Vol. 118, Issue 2
Figures / Tables found in this record: