
Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals
Abstract
In the past 30 years, Kohn–Sham density functional theory has emerged as the most popular electronic structure method in computational chemistry. To assess the ever-increasing number of approximate exchange-correlation functionals, this review benchmarks a total of 200 density functionals on a molecular database (MGCDB84) of nearly 5000 data points. The database employed, provided as Supplemental Data, is comprised of 84 data-sets and contains non-covalent interactions, isomerisation energies, thermochemistry, and barrier heights. In addition, the evolution of nonempirical and semi-empirical density functional design is reviewed, and guidelines are provided for the proper and effective use of density functionals. The most promising functional considered is ωB97M-V, a range-separated hybrid meta-GGA with VV10 nonlocal correlation, designed using a combinatorial approach. From the local GGAs, B97-D3, revPBE-D3, and BLYP-D3 are recommended, while from the local meta-GGAs, B97M-rV is the leading choice, followed by MS1-D3 and M06-LD3. The best hybrid GGAs are ωB97X-V, ωB97X-D3, and ωB97X-D, while useful hybrid meta-GGAs (besides ωB97M-V) include ωM05-D, M06-2X-D3, and MN15. Ultimately, today’s state-of-the-art functionals are close to achieving the level of accuracy desired for a broad range of chemical applications, and the principal remaining limitations are associated with systems that exhibit significant self-interaction/delocalisation errors and/or strong correlation effects.
- Authors:
-
- Kenneth S. Pitzer Center for Theoretical Chemistry, Department of Chemistry, University of California, Berkeley, CA, USA
- Kenneth S. Pitzer Center for Theoretical Chemistry, Department of Chemistry, University of California, Berkeley, CA, USA, Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
- Publication Date:
- Research Org.:
- Lawrence Berkeley National Laboratory (LBNL), Berkeley, CA (United States)
- Sponsoring Org.:
- USDOE Office of Science (SC), Basic Energy Sciences (BES)
- OSTI Identifier:
- 1510353
- Alternate Identifier(s):
- OSTI ID: 1477255
- Grant/Contract Number:
- AC02- 05CH11231; AC02-05CH11231
- Resource Type:
- Published Article
- Journal Name:
- Molecular Physics
- Additional Journal Information:
- Journal Name: Molecular Physics Journal Volume: 115 Journal Issue: 19; Journal ID: ISSN 0026-8976
- Publisher:
- Informa UK Limited
- Country of Publication:
- United Kingdom
- Language:
- English
- Subject:
- 37 INORGANIC, ORGANIC, PHYSICAL, AND ANALYTICAL CHEMISTRY; 74 ATOMIC AND MOLECULAR PHYSICS; Density functional theory; DFT; benchmark; chemistry database; density functionals
Citation Formats
Mardirossian, Narbe, and Head-Gordon, Martin. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. United Kingdom: N. p., 2017.
Web. doi:10.1080/00268976.2017.1333644.
Mardirossian, Narbe, & Head-Gordon, Martin. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. United Kingdom. https://doi.org/10.1080/00268976.2017.1333644
Mardirossian, Narbe, and Head-Gordon, Martin. Wed .
"Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals". United Kingdom. https://doi.org/10.1080/00268976.2017.1333644.
@article{osti_1510353,
title = {Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals},
author = {Mardirossian, Narbe and Head-Gordon, Martin},
abstractNote = {In the past 30 years, Kohn–Sham density functional theory has emerged as the most popular electronic structure method in computational chemistry. To assess the ever-increasing number of approximate exchange-correlation functionals, this review benchmarks a total of 200 density functionals on a molecular database (MGCDB84) of nearly 5000 data points. The database employed, provided as Supplemental Data, is comprised of 84 data-sets and contains non-covalent interactions, isomerisation energies, thermochemistry, and barrier heights. In addition, the evolution of nonempirical and semi-empirical density functional design is reviewed, and guidelines are provided for the proper and effective use of density functionals. The most promising functional considered is ωB97M-V, a range-separated hybrid meta-GGA with VV10 nonlocal correlation, designed using a combinatorial approach. From the local GGAs, B97-D3, revPBE-D3, and BLYP-D3 are recommended, while from the local meta-GGAs, B97M-rV is the leading choice, followed by MS1-D3 and M06-LD3. The best hybrid GGAs are ωB97X-V, ωB97X-D3, and ωB97X-D, while useful hybrid meta-GGAs (besides ωB97M-V) include ωM05-D, M06-2X-D3, and MN15. Ultimately, today’s state-of-the-art functionals are close to achieving the level of accuracy desired for a broad range of chemical applications, and the principal remaining limitations are associated with systems that exhibit significant self-interaction/delocalisation errors and/or strong correlation effects.},
doi = {10.1080/00268976.2017.1333644},
journal = {Molecular Physics},
number = 19,
volume = 115,
place = {United Kingdom},
year = {Wed Jun 21 00:00:00 EDT 2017},
month = {Wed Jun 21 00:00:00 EDT 2017}
}
https://doi.org/10.1080/00268976.2017.1333644
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
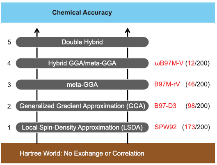 Figure 1.: Perdew’s metaphorical Jacob’s Ladder, composed of five rungs corresponding to increasingly sophisticated models for the unknown exchange-correlation functional of DFT. Since each rung contains new physical content that is missing in lower rungs, improved accuracy should be attainable at each higher level. This is illustrated by reporting themore »
Figure 1.: Perdew’s metaphorical Jacob’s Ladder, composed of five rungs corresponding to increasingly sophisticated models for the unknown exchange-correlation functional of DFT. Since each rung contains new physical content that is missing in lower rungs, improved accuracy should be attainable at each higher level. This is illustrated by reporting themore »
Works referenced in this record:
Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics
journal, March 2014
- Peverati, Roberto; Truhlar, Donald G.
- Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, Vol. 372, Issue 2011
Benchmark Interaction Energies for Biologically Relevant Noncovalent Complexes Containing Divalent Sulfur
journal, January 2012
- Mintz, Benjamin J.; Parks, Jerry M.
- The Journal of Physical Chemistry A, Vol. 116, Issue 3
An assessment of theoretical procedures for π -conjugation stabilisation energies in enones
journal, December 2014
- Yu, Li-Juan; Sarrami, Farzaneh; Karton, Amir
- Molecular Physics, Vol. 113, Issue 11
Evaluation of B3LYP, X3LYP, and M06-Class Density Functionals for Predicting the Binding Energies of Neutral, Protonated, and Deprotonated Water Clusters
journal, March 2009
- Bryantsev, Vyacheslav S.; Diallo, Mamadou S.; van Duin, Adri C. T.
- Journal of Chemical Theory and Computation, Vol. 5, Issue 4
MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids
journal, February 2016
- Yu, Haoyu S.; He, Xiao; Truhlar, Donald G.
- Journal of Chemical Theory and Computation, Vol. 12, Issue 3
Semiempirical hybrid density functional with perturbative second-order correlation
journal, January 2006
- Grimme, Stefan
- The Journal of Chemical Physics, Vol. 124, Issue 3
Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis
journal, August 1980
- Vosko, S. H.; Wilk, L.; Nusair, M.
- Canadian Journal of Physics, Vol. 58, Issue 8
Density functionals for static, dynamical, and strong correlation
journal, February 2013
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 138, Issue 7
Determination of Barrier Heights for Proton Exchange in Small Water, Ammonia, and Hydrogen Fluoride Clusters with G4(MP2)-Type, MP n , and SCS-MP n Procedures—A Caveat
journal, August 2012
- Karton, Amir; O’Reilly, Robert J.; Chan, Bun
- Journal of Chemical Theory and Computation, Vol. 8, Issue 9
Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials
journal, November 2001
- Wilson, Philip J.; Bradley, Thomas J.; Tozer, David J.
- The Journal of Chemical Physics, Vol. 115, Issue 20
Basis Set Convergence of the Post-CCSD(T) Contribution to Noncovalent Interaction Energies
journal, June 2014
- Smith, Daniel G. A.; Jankowski, Piotr; Slawik, Michał
- Journal of Chemical Theory and Computation, Vol. 10, Issue 8
Benchmark Thermochemistry of the C n H 2 n +2 Alkane Isomers ( n = 2−8) and Performance of DFT and Composite Ab Initio Methods for Dispersion-Driven Isomeric Equilibria
journal, July 2009
- Karton, Amir; Gruzman, David; Martin, Jan M. L.
- The Journal of Physical Chemistry A, Vol. 113, Issue 29
Global Hybrid Functionals: A Look at the Engine under the Hood
journal, November 2010
- Csonka, Gábor I.; Perdew, John P.; Ruzsinszky, Adrienn
- Journal of Chemical Theory and Computation, Vol. 6, Issue 12
A new mixing of Hartree–Fock and local density‐functional theories
journal, January 1993
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 98, Issue 2
Laplacian-level density functionals for the kinetic energy density and exchange-correlation energy
journal, April 2007
- Perdew, John P.; Constantin, Lucian A.
- Physical Review B, Vol. 75, Issue 15
The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals
journal, July 2007
- Zhao, Yan; Truhlar, Donald G.
- Theoretical Chemistry Accounts, Vol. 120, Issue 1-3
Accurate description of van der Waals complexes by density functional theory including empirical corrections
journal, January 2004
- Grimme, Stefan
- Journal of Computational Chemistry, Vol. 25, Issue 12
Benchmark Energetic Data in a Model System for Grubbs II Metathesis Catalysis and Their Use for the Development, Assessment, and Validation of Electronic Structure Methods
journal, January 2009
- Zhao, Yan; Truhlar, Donald G.
- Journal of Chemical Theory and Computation, Vol. 5, Issue 2
Advances in molecular quantum chemistry contained in the Q-Chem 4 program package
journal, September 2014
- Shao, Yihan; Gan, Zhengting; Epifanovsky, Evgeny
- Molecular Physics, Vol. 113, Issue 2
Orbital-free kinetic-energy density functionals with a density-dependent kernel
journal, December 1999
- Wang, Yan Alexander; Govind, Niranjan; Carter, Emily A.
- Physical Review B, Vol. 60, Issue 24
Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models
journal, January 1998
- Adamo, Carlo; Barone, Vincenzo
- The Journal of Chemical Physics, Vol. 108, Issue 2
Noncovalent Interactions in Specific Recognition Motifs of Protein–DNA Complexes
journal, January 2017
- Stasyuk, Olga A.; Jakubec, David; Vondrášek, Jiří
- Journal of Chemical Theory and Computation, Vol. 13, Issue 2
Improving the orbital-free density functional theory description of covalent materials
journal, January 2005
- Zhou, Baojing; Ligneres, Vincent L.; Carter, Emily A.
- The Journal of Chemical Physics, Vol. 122, Issue 4
Density-functional errors in ionization potential with increasing system size
journal, May 2015
- Whittleton, Sarah R.; Sosa Vazquez, Xochitl A.; Isborn, Christine M.
- The Journal of Chemical Physics, Vol. 142, Issue 18
Density functional calculations of molecular bond energies
journal, April 1986
- Becke, A. D.
- The Journal of Chemical Physics, Vol. 84, Issue 8
A study of the Xαβ-exchange approximation on atoms for use in molecular and solid-state calculations
journal, January 1975
- Schwarz, Karlheinz
- Chemical Physics, Vol. 7, Issue 1
Exchange–Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient
journal, June 2012
- Peverati, Roberto; Truhlar, Donald G.
- Journal of Chemical Theory and Computation, Vol. 8, Issue 7
Thermochemical benchmarking of hydrocarbon bond separation reaction energies: Jacob's ladder is not reversed!
journal, September 2010
- Krieg, Helge; Grimme, Stefan
- Molecular Physics, Vol. 108, Issue 19-20
Effect of the damping function in dispersion corrected density functional theory
journal, March 2011
- Grimme, Stefan; Ehrlich, Stephan; Goerigk, Lars
- Journal of Computational Chemistry, Vol. 32, Issue 7
Basis set limit coupled-cluster studies of hydrogen-bonded systems
journal, February 2015
- Boese, A. Daniel
- Molecular Physics, Vol. 113, Issue 13-14
Improved Statistical Exchange Approximation for Inhomogeneous Many-Electron Systems
journal, April 1969
- Herman, Frank; Van Dyke, John P.; Ortenburger, Irene B.
- Physical Review Letters, Vol. 22, Issue 16
Treating London-Dispersion Effects with the Latest Minnesota Density Functionals: Problems and Possible Solutions
journal, September 2015
- Goerigk, Lars
- The Journal of Physical Chemistry Letters, Vol. 6, Issue 19
Representative Benchmark Suites for Barrier Heights of Diverse Reaction Types and Assessment of Electronic Structure Methods for Thermochemical Kinetics
journal, December 2006
- Zheng, Jingjing; Zhao, Yan; Truhlar, Donald G.
- Journal of Chemical Theory and Computation, Vol. 3, Issue 2
Density‐functional thermochemistry. II. The effect of the Perdew–Wang generalized‐gradient correlation correction
journal, December 1992
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 97, Issue 12
Density Functional Theory and Hydrogen Bonds: Are We There Yet?
journal, February 2015
- Boese, A. Daniel
- ChemPhysChem, Vol. 16, Issue 5
Development of density functionals for thermochemical kinetics
journal, August 2004
- Boese, A. Daniel; Martin, Jan M. L.
- The Journal of Chemical Physics, Vol. 121, Issue 8
Performance of the van der Waals Density Functional VV10 and (hybrid)GGA Variants for Thermochemistry and Noncovalent Interactions
journal, October 2011
- Hujo, Waldemar; Grimme, Stefan
- Journal of Chemical Theory and Computation, Vol. 7, Issue 12
Benchmark ab Initio Conformational Energies for the Proteinogenic Amino Acids through Explicitly Correlated Methods. Assessment of Density Functional Methods
journal, December 2015
- Kesharwani, Manoj K.; Karton, Amir; Martin, Jan M. L.
- Journal of Chemical Theory and Computation, Vol. 12, Issue 1
Real-space post-Hartree–Fock correlation models
journal, February 2005
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 122, Issue 6
Communication: Two-determinant mixing with a strong-correlation density functional
journal, July 2013
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 139, Issue 2
Perspective: Fifty years of density-functional theory in chemical physics
journal, May 2014
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 140, Issue 18
Coulomb-attenuated exchange energy density functionals
journal, July 1996
- Gill, Peter M. W.; Adamson, Ross D.; Pople, John A.
- Molecular Physics, Vol. 88, Issue 4
A semiempirical generalized gradient approximation exchange-correlation functional
journal, September 2004
- Keal, Thomas W.; Tozer, David J.
- The Journal of Chemical Physics, Vol. 121, Issue 12
A new one-parameter progressive Colle–Salvetti-type correlation functional
journal, June 1999
- Tsuneda, Takao; Suzumura, Toshihisa; Hirao, Kimihiko
- The Journal of Chemical Physics, Vol. 110, Issue 22
A Simplification of the Hartree-Fock Method
journal, February 1951
- Slater, J. C.
- Physical Review, Vol. 81, Issue 3
Extensions of the S66 Data Set: More Accurate Interaction Energies and Angular-Displaced Nonequilibrium Geometries
journal, October 2011
- Řezáč, Jan; Riley, Kevin E.; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 7, Issue 11
Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces
journal, April 2008
- Perdew, John P.; Ruzsinszky, Adrienn; Csonka, Gábor I.
- Physical Review Letters, Vol. 100, Issue 13
Mapping the genome of meta-generalized gradient approximation density functionals: The search for B97M-V
journal, February 2015
- Mardirossian, Narbe; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 142, Issue 7
Multi-coefficient extrapolated density functional theory for thermochemistry and thermochemical kinetics
journal, January 2005
- Zhao, Yan; Lynch, Benjamin J.; Truhlar, Donald G.
- Physical Chemistry Chemical Physics, Vol. 7, Issue 1
Do Practical Standard Coupled Cluster Calculations Agree Better than Kohn–Sham Calculations with Currently Available Functionals When Compared to the Best Available Experimental Data for Dissociation Energies of Bonds to 3 d Transition Metals?
journal, April 2015
- Xu, Xuefei; Zhang, Wenjing; Tang, Mingsheng
- Journal of Chemical Theory and Computation, Vol. 11, Issue 5
Accurate Description of Intermolecular Interactions Involving Ions Using Symmetry-Adapted Perturbation Theory
journal, May 2015
- Lao, Ka Un; Schäffer, Rainer; Jansen, Georg
- Journal of Chemical Theory and Computation, Vol. 11, Issue 6
Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions
journal, August 2004
- Zhao, Yan; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 108, Issue 33
The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors
journal, October 1970
- Boys, S.F.; Bernardi, F.
- Molecular Physics, Vol. 19, Issue 4, p. 553-566
The exchange-correlation potential in Kohn–Sham nuclear magnetic resonance shielding calculations
journal, August 2003
- Keal, Thomas W.; Tozer, David J.
- The Journal of Chemical Physics, Vol. 119, Issue 6
Reformulation of the D3(Becke–Johnson) Dispersion Correction without Resorting to Higher than C 6 Dispersion Coefficients
journal, June 2015
- Schröder, Heiner; Creon, Anne; Schwabe, Tobias
- Journal of Chemical Theory and Computation, Vol. 11, Issue 7
Local Scaling Correction for Reducing Delocalization Error in Density Functional Approximations
journal, February 2015
- Li, Chen; Zheng, Xiao; Cohen, Aron J.
- Physical Review Letters, Vol. 114, Issue 5
Formal Estimation of Errors in Computed Absolute Interaction Energies of Protein−Ligand Complexes
journal, February 2011
- Faver, John C.; Benson, Mark L.; He, Xiao
- Journal of Chemical Theory and Computation, Vol. 7, Issue 3
Analysis of the Electronic Exchange in Atoms
journal, February 1972
- Lindgren, Ingvar; Schwarz, Karlheinz
- Physical Review A, Vol. 5, Issue 2
How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements?
journal, August 2016
- Mardirossian, Narbe; Head-Gordon, Martin
- Journal of Chemical Theory and Computation, Vol. 12, Issue 9
Communication: Self-interaction correction with unitary invariance in density functional theory
journal, March 2014
- Pederson, Mark R.; Ruzsinszky, Adrienn; Perdew, John P.
- The Journal of Chemical Physics, Vol. 140, Issue 12
Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions
journal, December 2010
- Goerigk, Lars; Grimme, Stefan
- Journal of Chemical Theory and Computation, Vol. 7, Issue 2
Accurate and Efficient Quantum Chemistry Calculations for Noncovalent Interactions in Many-Body Systems: The XSAPT Family of Methods
journal, December 2014
- Lao, Ka Un; Herbert, John M.
- The Journal of Physical Chemistry A, Vol. 119, Issue 2
Exploring the limit of accuracy for density functionals based on the generalized gradient approximation: Local, global hybrid, and range-separated hybrid functionals with and without dispersion corrections
journal, May 2014
- Mardirossian, Narbe; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 140, Issue 18
The surface energy of a bounded electron gas
journal, August 1974
- Harris, J.; Jones, R. O.
- Journal of Physics F: Metal Physics, Vol. 4, Issue 8
Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States
journal, December 2006
- Zhao, Yan; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 110, Issue 49
Standard grids for high-precision integration of modern density functionals: SG-2 and SG-3
journal, February 2017
- Dasgupta, Saswata; Herbert, John M.
- Journal of Computational Chemistry, Vol. 38, Issue 12
An improved and broadly accurate local approximation to the exchange–correlation density functional: The MN12-L functional for electronic structure calculations in chemistry and physics
journal, January 2012
- Peverati, Roberto; Truhlar, Donald G.
- Physical Chemistry Chemical Physics, Vol. 14, Issue 38
An investigation of the performance of a hybrid of Hartree-Fock and density functional theory
journal, March 1992
- Gill, Peter M. W.; Johnson, Benny G.; Pople, John A.
- International Journal of Quantum Chemistry, Vol. 44, Issue S26
Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs
journal, January 2006
- Jurečka, Petr; Šponer, Jiří; Černý, Jiří
- Physical Chemistry Chemical Physics, Vol. 8, Issue 17, p. 1985-1993
Benchmark Calculations of Noncovalent Interactions of Halogenated Molecules
journal, September 2012
- Řezáč, Jan; Riley, Kevin E.; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 8, Issue 11
Oscillations in meta-generalized-gradient approximation potential energy surfaces for dispersion-bound complexes
journal, July 2009
- Johnson, Erin R.; Becke, Axel D.; Sherrill, C. David
- The Journal of Chemical Physics, Vol. 131, Issue 3
Hartree-Fock exchange energy of an inhomogeneous electron gas
journal, June 1983
- Becke, A. D.
- International Journal of Quantum Chemistry, Vol. 23, Issue 6
Communication: A new class of non-empirical explicit density functionals on the third rung of Jacob’s ladder
journal, September 2015
- de Silva, Piotr; Corminboeuf, Clémence
- The Journal of Chemical Physics, Vol. 143, Issue 11
What Can We Learn about Dispersion from the Conformer Surface of n -Pentane?
journal, March 2013
- Martin, Jan M. L.
- The Journal of Physical Chemistry A, Vol. 117, Issue 14
Adiabatic Connection for Kinetics
journal, June 2000
- Lynch, Benjamin J.; Fast, Patton L.; Harris, Maegan
- The Journal of Physical Chemistry A, Vol. 104, Issue 21
Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory
journal, March 2009
- Stein, Tamar; Kronik, Leeor; Baer, Roi
- Journal of the American Chemical Society, Vol. 131, Issue 8
A Systematic CCSD(T) Study of Long-Range and Noncovalent Interactions between Benzene and a Series of First- and Second-Row Hydrides and Rare Gas Atoms
journal, February 2009
- Crittenden, Deborah L.
- The Journal of Physical Chemistry A, Vol. 113, Issue 8
Bond Dissociation Energies for Diatomic Molecules Containing 3d Transition Metals: Benchmark Scalar-Relativistic Coupled-Cluster Calculations for 20 Molecules
journal, February 2017
- Cheng, Lan; Gauss, Jürgen; Ruscic, Branko
- Journal of Chemical Theory and Computation, Vol. 13, Issue 3
A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP)
journal, July 2004
- Yanai, Takeshi; Tew, David P.; Handy, Nicholas C.
- Chemical Physics Letters, Vol. 393, Issue 1-3, p. 51-57
Prediction of Bond Dissociation Energies/Heats of Formation for Diatomic Transition Metal Compounds: CCSD(T) Works
journal, January 2017
- Fang, Zongtang; Vasiliu, Monica; Peterson, Kirk A.
- Journal of Chemical Theory and Computation, Vol. 13, Issue 3
Benchmark tests of a strongly constrained semilocal functional with a long-range dispersion correction
journal, September 2016
- Brandenburg, J. G.; Bates, J. E.; Sun, J.
- Physical Review B, Vol. 94, Issue 11
An improved treatment of empirical dispersion and a many-body energy decomposition scheme for the explicit polarization plus symmetry-adapted perturbation theory (XSAPT) method
journal, July 2013
- Lao, Ka Un; Herbert, John M.
- The Journal of Chemical Physics, Vol. 139, Issue 3
Correlation energies in the spin-density functional formalism: II. Applications and empirical corrections
journal, January 1980
- Stoll, Hermann; Golka, Elisabeth; Preu�, Heinzwerner
- Theoretica Chimica Acta, Vol. 55, Issue 1
The van der Waals potentials between all the rare gas atoms from He to Rn
journal, March 2003
- Tang, K. T.; Toennies, J. P.
- The Journal of Chemical Physics, Vol. 118, Issue 11
Density‐functional thermochemistry. III. The role of exact exchange
journal, April 1993
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 98, Issue 7, p. 5648-5652
The Mechanism of Dihydrogen Activation by Frustrated Lewis Pairs Revisited
journal, January 2010
- Grimme, Stefan; Kruse, Holger; Goerigk, Lars
- Angewandte Chemie International Edition, Vol. 49, Issue 8
Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules
journal, November 2005
- Dreuw, Andreas; Head-Gordon, Martin
- Chemical Reviews, Vol. 105, Issue 11
Revealing Noncovalent Interactions
journal, May 2010
- Johnson, Erin R.; Keinan, Shahar; Mori-Sánchez, Paula
- Journal of the American Chemical Society, Vol. 132, Issue 18
Generalized Gradient Approximation That Recovers the Second-Order Density-Gradient Expansion with Optimized Across-the-Board Performance
journal, July 2011
- Peverati, Roberto; Zhao, Yan; Truhlar, Donald G.
- The Journal of Physical Chemistry Letters, Vol. 2, Issue 16
Higher-accuracy van der Waals density functional
journal, August 2010
- Lee, Kyuho; Murray, Éamonn D.; Kong, Lingzhu
- Physical Review B, Vol. 82, Issue 8
Assessment of theoretical procedures for a diverse set of isomerization reactions involving double-bond migration in conjugated dienes
journal, September 2014
- Yu, Li-Juan; Karton, Amir
- Chemical Physics, Vol. 441
Density-functional approximation for the correlation energy of the inhomogeneous electron gas
journal, June 1986
- Perdew, John P.
- Physical Review B, Vol. 33, Issue 12
High-level ab initio calculations for the four low-lying families of minima of (H[sub 2]O)[sub 20]. I. Estimates of MP2/CBS binding energies and comparison with empirical potentials
journal, January 2004
- Fanourgakis, George S.; Aprà, Edoardo; Xantheas, Sotiris S.
- The Journal of Chemical Physics, Vol. 121, Issue 6
A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions
journal, November 2006
- Zhao, Yan; Truhlar, Donald G.
- The Journal of Chemical Physics, Vol. 125, Issue 19, Article No. 194101
Many-electron self-interaction error in approximate density functionals
journal, November 2006
- Mori-Sánchez, Paula; Cohen, Aron J.; Yang, Weitao
- The Journal of Chemical Physics, Vol. 125, Issue 20
Assessment of Coupled Cluster Theory and more Approximate Methods for Hydrogen Bonded Systems
journal, September 2013
- Boese, A. Daniel
- Journal of Chemical Theory and Computation, Vol. 9, Issue 10
Toward reliable density functional methods without adjustable parameters: The PBE0 model
journal, April 1999
- Adamo, Carlo; Barone, Vincenzo
- The Journal of Chemical Physics, Vol. 110, Issue 13
Highly Accurate First-Principles Benchmark Data Sets for the Parametrization and Validation of Density Functional and Other Approximate Methods. Derivation of a Robust, Generally Applicable, Double-Hybrid Functional for Thermochemistry and Thermochemical Kinetics †
journal, December 2008
- Karton, Amir; Tarnopolsky, Alex; Lamère, Jean-François
- The Journal of Physical Chemistry A, Vol. 112, Issue 50
Atomic orbital basis sets: Atomic orbital basis sets
journal, October 2012
- Jensen, Frank
- Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 3, Issue 3
Can orbital-free density functional theory simulate molecules?
journal, February 2012
- Xia, Junchao; Huang, Chen; Shin, Ilgyou
- The Journal of Chemical Physics, Vol. 136, Issue 8
Inhomogeneous Electron Gas
journal, November 1964
- Hohenberg, P.; Kohn, W.
- Physical Review, Vol. 136, Issue 3B, p. B864-B871
Avoiding singularity problems associated with meta-GGA (generalized gradient approximation) exchange and correlation functionals containing the kinetic energy density
journal, December 2007
- Gräfenstein, Jürgen; Izotov, Dmitry; Cremer, Dieter
- The Journal of Chemical Physics, Vol. 127, Issue 21
Describing Noncovalent Interactions beyond the Common Approximations: How Accurate Is the “Gold Standard,” CCSD(T) at the Complete Basis Set Limit?
journal, April 2013
- Řezáč, Jan; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 9, Issue 5
The Performance of Density Functionals for Sulfate–Water Clusters
journal, February 2013
- Mardirossian, Narbe; Lambrecht, Daniel S.; McCaslin, Laura
- Journal of Chemical Theory and Computation, Vol. 9, Issue 3
Gaussian‐2 theory for molecular energies of first‐ and second‐row compounds
journal, June 1991
- Curtiss, Larry A.; Raghavachari, Krishnan; Trucks, Gary W.
- The Journal of Chemical Physics, Vol. 94, Issue 11
On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors
journal, October 2013
- Bauzá, Antonio; Alkorta, Ibon; Frontera, Antonio
- Journal of Chemical Theory and Computation, Vol. 9, Issue 11
ω B97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation
journal, June 2016
- Mardirossian, Narbe; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 144, Issue 21
Simulation of delocalized exchange by local density functionals
journal, March 2000
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 112, Issue 9
Hydrocarbon/Water Interactions: Encouraging Energetics and Structures from DFT but Disconcerting Discrepancies for Hessian Indices
journal, April 2012
- Copeland, Kari L.; Tschumper, Gregory S.
- Journal of Chemical Theory and Computation, Vol. 8, Issue 5
Benchmark Calculations of Three-Body Intermolecular Interactions and the Performance of Low-Cost Electronic Structure Methods
journal, June 2015
- Řezáč, Jan; Huang, Yuanhang; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 11, Issue 7
Density‐functional thermochemistry. I. The effect of the exchange‐only gradient correction
journal, February 1992
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 96, Issue 3
Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets
journal, June 2009
- Jain, Rupal; Bally, Thomas; Rablen, Paul R.
- The Journal of Organic Chemistry, Vol. 74, Issue 11
Polarizable interaction potential for water from coupled cluster calculations. I. Analysis of dimer potential energy surface
journal, March 2008
- Bukowski, Robert; Szalewicz, Krzysztof; Groenenboom, Gerrit C.
- The Journal of Chemical Physics, Vol. 128, Issue 9
Assessment of Theoretical Procedures for Calculating Barrier Heights for a Diverse Set of Water-Catalyzed Proton-Transfer Reactions
journal, April 2012
- Karton, Amir; O’Reilly, Robert J.; Radom, Leo
- The Journal of Physical Chemistry A, Vol. 116, Issue 16
Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals
journal, November 1997
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 107, Issue 20
A standard grid for density functional calculations
journal, July 1993
- Gill, Peter M. W.; Johnson, Benny G.; Pople, John A.
- Chemical Physics Letters, Vol. 209, Issue 5-6
A novel form for the exchange-correlation energy functional
journal, July 1998
- Van Voorhis, Troy; Scuseria, Gustavo E.
- The Journal of Chemical Physics, Vol. 109, Issue 2
Long-Range-Corrected Hybrids Based on a New Model Exchange Hole
journal, March 2009
- Weintraub, Elon; Henderson, Thomas M.; Scuseria, Gustavo E.
- Journal of Chemical Theory and Computation, Vol. 5, Issue 4
Consistent structures and interactions by density functional theory with small atomic orbital basis sets
journal, August 2015
- Grimme, Stefan; Brandenburg, Jan Gerit; Bannwarth, Christoph
- The Journal of Chemical Physics, Vol. 143, Issue 5
Conformers of Gaseous Cysteine
journal, May 2009
- Wilke, Jeremiah J.; Lind, Maria C.; Schaefer, Henry F.
- Journal of Chemical Theory and Computation, Vol. 5, Issue 6
Generalized Gradient Approximation Made Simple
journal, October 1996
- Perdew, John P.; Burke, Kieron; Ernzerhof, Matthias
- Physical Review Letters, Vol. 77, Issue 18, p. 3865-3868
Current- and spin-density-functional theory for inhomogeneous electronic systems in strong magnetic fields
journal, June 1988
- Vignale, G.; Rasolt, Mark
- Physical Review B, Vol. 37, Issue 18
A General Database for Main Group Thermochemistry, Kinetics, and Noncovalent Interactions − Assessment of Common and Reparameterized ( meta -)GGA Density Functionals
journal, November 2009
- Goerigk, Lars; Grimme, Stefan
- Journal of Chemical Theory and Computation, Vol. 6, Issue 1
Nonlocal van der Waals density functional made simple and efficient
journal, January 2013
- Sabatini, Riccardo; Gorni, Tommaso; de Gironcoli, Stefano
- Physical Review B, Vol. 87, Issue 4
Binding in Radical-Solvent Binary Complexes: Benchmark Energies and Performance of Approximate Methods
journal, February 2013
- Tentscher, Peter R.; Arey, J. Samuel
- Journal of Chemical Theory and Computation, Vol. 9, Issue 3
Explicitly correlated benchmark calculations on C 8 H 8 isomer energy separations: how accurate are DFT, double-hybrid, and composite ab initio procedures?
journal, April 2012
- Karton, Amir; Martin, Jan M. L.
- Molecular Physics, Vol. 110, Issue 19-20
Systematic optimization of long-range corrected hybrid density functionals
journal, February 2008
- Chai, Jeng-Da; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 128, Issue 8
Time-dependent density functional theory: Past, present, and future
journal, August 2005
- Burke, Kieron; Werschnik, Jan; Gross, E. K. U.
- The Journal of Chemical Physics, Vol. 123, Issue 6
Integration Grid Errors for Meta-GGA-Predicted Reaction Energies: Origin of Grid Errors for the M06 Suite of Functionals
journal, January 2010
- Wheeler, Steven E.; Houk, K. N.
- Journal of Chemical Theory and Computation, Vol. 6, Issue 2
The Melatonin Conformer Space: Benchmark and Assessment of Wave Function and DFT Methods for a Paradigmatic Biological and Pharmacological Molecule
journal, March 2013
- Fogueri, Uma R.; Kozuch, Sebastian; Karton, Amir
- The Journal of Physical Chemistry A, Vol. 117, Issue 10
W4-11: A high-confidence benchmark dataset for computational thermochemistry derived from first-principles W4 data
journal, July 2011
- Karton, Amir; Daon, Shauli; Martin, Jan M. L.
- Chemical Physics Letters, Vol. 510, Issue 4-6
Optimized statistical exchange parameters ? for atoms with higherZ
journal, January 1974
- Schwarz, Karlheinz
- Theoretica Chimica Acta, Vol. 34, Issue 3
Communication: Effect of the orbital-overlap dependence in the meta generalized gradient approximation
journal, August 2012
- Sun, Jianwei; Xiao, Bing; Ruzsinszky, Adrienn
- The Journal of Chemical Physics, Vol. 137, Issue 5
Explicitly correlated coupled cluster benchmarks with realistic-sized ligands for some late-transition metal reactions: basis sets convergence and performance of more approximate methods
journal, February 2014
- Kesharwani, Manoj K.; Martin, Jan M. L.
- Theoretical Chemistry Accounts, Vol. 133, Issue 3
Meta-GGA Exchange-Correlation Functional with a Balanced Treatment of Nonlocality
journal, April 2013
- Constantin, Lucian A.; Fabiano, E.; Della Sala, F.
- Journal of Chemical Theory and Computation, Vol. 9, Issue 5
Construction of a generalized gradient approximation by restoring the density-gradient expansion and enforcing a tight Lieb–Oxford bound
journal, May 2008
- Zhao, Yan; Truhlar, Donald G.
- The Journal of Chemical Physics, Vol. 128, Issue 18
Multiconfiguration Pair-Density Functional Theory: A New Way To Treat Strongly Correlated Systems
journal, December 2016
- Gagliardi, Laura; Truhlar, Donald G.; Li Manni, Giovanni
- Accounts of Chemical Research, Vol. 50, Issue 1
Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory
journal, May 2016
- Smith, Daniel G. A.; Burns, Lori A.; Patkowski, Konrad
- The Journal of Physical Chemistry Letters, Vol. 7, Issue 12
A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions
journal, January 2011
- Goerigk, Lars; Grimme, Stefan
- Physical Chemistry Chemical Physics, Vol. 13, Issue 14
Push it to the limit: Characterizing the convergence of common sequences of basis sets for intermolecular interactions as described by density functional theory
journal, May 2016
- Witte, Jonathon; Neaton, Jeffrey B.; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 144, Issue 19
New exchange-correlation density functionals: The role of the kinetic-energy density
journal, June 2002
- Boese, A. Daniel; Handy, Nicholas C.
- The Journal of Chemical Physics, Vol. 116, Issue 22
Benchmark Database of Barrier Heights for Heavy Atom Transfer, Nucleophilic Substitution, Association, and Unimolecular Reactions and Its Use to Test Theoretical Methods
journal, March 2005
- Zhao, Yan; González-García, Núria; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 109, Issue 9
New generalized gradient approximation functionals
journal, January 2000
- Boese, A. Daniel; Doltsinis, Nikos L.; Handy, Nicholas C.
- The Journal of Chemical Physics, Vol. 112, Issue 4
Density-functional approaches to noncovalent interactions: A comparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM) theory, and specialized functionals
journal, February 2011
- Burns, Lori A.; Mayagoitia, Álvaro Vázquez-; Sumpter, Bobby G.
- The Journal of Chemical Physics, Vol. 134, Issue 8
Semilocal and hybrid meta-generalized gradient approximations based on the understanding of the kinetic-energy-density dependence
journal, January 2013
- Sun, Jianwei; Haunschild, Robin; Xiao, Bing
- The Journal of Chemical Physics, Vol. 138, Issue 4
Density functional theory is straying from the path toward the exact functional
journal, January 2017
- Medvedev, Michael G.; Bushmarinov, Ivan S.; Sun, Jianwei
- Science, Vol. 355, Issue 6320
What Are the Ground State Structures of C 20 and C 24 ? An Explicitly Correlated Ab Initio Approach
journal, December 2015
- Manna, Debashree; Martin, Jan M. L.
- The Journal of Physical Chemistry A, Vol. 120, Issue 1
Strong Correlation in Kohn-Sham Density Functional Theory
journal, December 2012
- Malet, Francesc; Gori-Giorgi, Paola
- Physical Review Letters, Vol. 109, Issue 24
Benchmark Structures and Binding Energies of Small Water Clusters with Anharmonicity Corrections
journal, November 2011
- Temelso, Berhane; Archer, Kaye A.; Shields, George C.
- The Journal of Physical Chemistry A, Vol. 115, Issue 43
Performance of Ab Initio and Density Functional Methods for Conformational Equilibria of C n H 2 n +2 Alkane Isomers ( n = 4−8) †
journal, October 2009
- Gruzman, David; Karton, Amir; Martin, Jan M. L.
- The Journal of Physical Chemistry A, Vol. 113, Issue 43
Reaction barrier heights for cycloreversion of heterocyclic rings: An Achilles’ heel for DFT and standard ab initio procedures
journal, September 2015
- Yu, Li-Juan; Sarrami, Farzaneh; O’Reilly, Robert J.
- Chemical Physics, Vol. 458
Halogen Bonding from Dispersion-Corrected Density-Functional Theory: The Role of Delocalization Error
journal, November 2014
- Otero-de-la-Roza, A.; Johnson, Erin R.; DiLabio, Gino A.
- Journal of Chemical Theory and Computation, Vol. 10, Issue 12
Density-functional theory in strong magnetic fields
journal, November 1987
- Vignale, G.; Rasolt, Mark
- Physical Review Letters, Vol. 59, Issue 20
Nonseparable exchange–correlation functional for molecules, including homogeneous catalysis involving transition metals
journal, January 2015
- Yu, Haoyu S.; Zhang, Wenjing; Verma, Pragya
- Physical Chemistry Chemical Physics, Vol. 17, Issue 18
Density-gradient analysis for density functional theory: Application to atoms
journal, January 1997
- Zupan, Ale?; Perdew, John P.; Burke, Kieron
- International Journal of Quantum Chemistry, Vol. 61, Issue 5
Double-hybrid density functionals with long-range dispersion corrections: higher accuracy and extended applicability
journal, January 2007
- Schwabe, Tobias; Grimme, Stefan
- Physical Chemistry Chemical Physics, Vol. 9, Issue 26
A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu
journal, April 2010
- Grimme, Stefan; Antony, Jens; Ehrlich, Stephan
- The Journal of Chemical Physics, Vol. 132, Issue 15
Self-Consistent Equations Including Exchange and Correlation Effects
journal, November 1965
- Kohn, W.; Sham, L. J.
- Physical Review, Vol. 140, Issue 4A, p. A1133-A1138
Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections
journal, January 2008
- Chai, Jeng-Da; Head-Gordon, Martin
- Physical Chemistry Chemical Physics, Vol. 10, Issue 44
Semiempirical hybrid functional with improved performance in an extensive chemical assessment
journal, September 2005
- Keal, Thomas W.; Tozer, David J.
- The Journal of Chemical Physics, Vol. 123, Issue 12
A method for improving the physical realism of first-principles band structure calculations
journal, January 1969
- Herman, F.; Ortenburger, I. B.; Van Dyke, J. P.
- International Journal of Quantum Chemistry, Vol. 4, Issue S3B
Beyond Energies: Geometries of Nonbonded Molecular Complexes as Metrics for Assessing Electronic Structure Approaches
journal, March 2015
- Witte, Jonathon; Goldey, Matthew; Neaton, Jeffrey B.
- Journal of Chemical Theory and Computation, Vol. 11, Issue 4
S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures
journal, July 2011
- Řezáč, Jan; Riley, Kevin E.; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 7, Issue 8
New accurate benchmark energies for large water clusters: DFT is better than expected
journal, January 2014
- Anacker, Tony; Friedrich, Joachim
- Journal of Computational Chemistry, Vol. 35, Issue 8
A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states
journal, February 2009
- Rohrdanz, Mary A.; Martins, Katie M.; Herbert, John M.
- The Journal of Chemical Physics, Vol. 130, Issue 5
Performance of spin-component-scaled Møller–Plesset theory (SCS-MP2) for potential energy curves of noncovalent interactions
journal, January 2007
- Takatani, Tait; David Sherrill, C.
- Physical Chemistry Chemical Physics, Vol. 9, Issue 46
Combining long-range configuration interaction with short-range density functionals
journal, August 1997
- Leininger, Thierry; Stoll, Hermann; Werner, Hans-Joachim
- Chemical Physics Letters, Vol. 275, Issue 3-4
Can DFT and ab initio methods describe all aspects of the potential energy surface of cycloreversion reactions?
journal, September 2015
- Yu, Li-Juan; Sarrami, Farzaneh; O'Reilly, Robert J.
- Molecular Physics, Vol. 114, Issue 1
Halogen Bonds: Benchmarks and Theoretical Analysis
journal, March 2013
- Kozuch, Sebastian; Martin, Jan M. L.
- Journal of Chemical Theory and Computation, Vol. 9, Issue 4
Quantifying the effects of the self-interaction error in DFT: When do the delocalized states appear?
journal, June 2005
- Lundberg, Marcus; Siegbahn, Per E. M.
- The Journal of Chemical Physics, Vol. 122, Issue 22
Dispersion-Corrected Mean-Field Electronic Structure Methods
journal, April 2016
- Grimme, Stefan; Hansen, Andreas; Brandenburg, Jan Gerit
- Chemical Reviews, Vol. 116, Issue 9
Effectiveness of Diffuse Basis Functions for Calculating Relative Energies by Density Functional Theory
journal, March 2003
- Lynch, Benjamin J.; Zhao, Yan; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 107, Issue 9
Effects of Heteroatoms on Aromatic π−π Interactions: Benzene−Pyridine and Pyridine Dimer
journal, February 2009
- Hohenstein, Edward G.; Sherrill, C. David
- The Journal of Physical Chemistry A, Vol. 113, Issue 5
Van der Waals Density Functional for General Geometries
journal, June 2004
- Dion, M.; Rydberg, H.; Schröder, E.
- Physical Review Letters, Vol. 92, Issue 24
Current-density dependent exchange-correlation functionals
journal, June 1996
- Becke, Axel D.
- Canadian Journal of Chemistry, Vol. 74, Issue 6
A dataset of highly accurate homolytic NBr bond dissociation energies obtained by Means of W2 theory
journal, October 2015
- O'Reilly, Robert J.; Karton, Amir
- International Journal of Quantum Chemistry, Vol. 116, Issue 1
Exchange-correlation functional with broad accuracy for metallic and nonmetallic compounds, kinetics, and noncovalent interactions
journal, October 2005
- Zhao, Yan; Schultz, Nathan E.; Truhlar, D. G.
- The Journal of Chemical Physics, Vol. 123, Issue 16
Density functional theory with London dispersion corrections: Density functional theory with London dispersion corrections
journal, March 2011
- Grimme, Stefan
- Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 1, Issue 2
Semiempirical GGA-type density functional constructed with a long-range dispersion correction
journal, January 2006
- Grimme, Stefan
- Journal of Computational Chemistry, Vol. 27, Issue 15, p. 1787-1799
Density‐functional thermochemistry. IV. A new dynamical correlation functional and implications for exact‐exchange mixing
journal, January 1996
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 104, Issue 3
Why are the Interaction Energies of Charge-Transfer Complexes Challenging for DFT?
journal, April 2012
- Steinmann, Stephan N.; Piemontesi, Cyril; Delachat, Aurore
- Journal of Chemical Theory and Computation, Vol. 8, Issue 5
Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes
journal, December 2003
- Staroverov, Viktor N.; Scuseria, Gustavo E.; Tao, Jianmin
- The Journal of Chemical Physics, Vol. 119, Issue 23
Semilocal density functional obeying a strongly tightened bound for exchange
journal, January 2015
- Sun, Jianwei; Perdew, John P.; Ruzsinszky, Adrienn
- Proceedings of the National Academy of Sciences, Vol. 112, Issue 3
Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation
journal, October 2011
- Peverati, Roberto; Truhlar, Donald G.
- The Journal of Physical Chemistry Letters, Vol. 2, Issue 21
Property-optimized Gaussian basis sets for molecular response calculations
journal, October 2010
- Rappoport, Dmitrij; Furche, Filipp
- The Journal of Chemical Physics, Vol. 133, Issue 13
Accurate reaction barrier heights of pericyclic reactions: Surprisingly large deviations for the CBS-QB3 composite method and their consequences in DFT benchmark studies
journal, February 2015
- Karton, Amir; Goerigk, Lars
- Journal of Computational Chemistry, Vol. 36, Issue 9
Nonlocal van der Waals density functional: The simpler the better
journal, December 2010
- Vydrov, Oleg A.; Van Voorhis, Troy
- The Journal of Chemical Physics, Vol. 133, Issue 24
Optimization of the Statistical Exchange Parameter for the Free Atoms H through Nb
journal, April 1972
- Schwarz, Karlheinz
- Physical Review B, Vol. 5, Issue 7
Development and Assessment of a New Hybrid Density Functional Model for Thermochemical Kinetics
journal, April 2004
- Zhao, Yan; Lynch, Benjamin J.; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 108, Issue 14
Communication: A global hybrid generalized gradient approximation to the exchange-correlation functional that satisfies the second-order density-gradient constraint and has broad applicability in chemistry
journal, November 2011
- Peverati, Roberto; Truhlar, Donald G.
- The Journal of Chemical Physics, Vol. 135, Issue 19
Basis set convergence of the coupled-cluster correction, δMP2CCSD(T): Best practices for benchmarking non-covalent interactions and the attendant revision of the S22, NBC10, HBC6, and HSG databases
journal, November 2011
- Marshall, Michael S.; Burns, Lori A.; Sherrill, C. David
- The Journal of Chemical Physics, Vol. 135, Issue 19
A new inhomogeneity parameter in density-functional theory
journal, August 1998
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 109, Issue 6
mBEEF: An accurate semi-local Bayesian error estimation density functional
journal, April 2014
- Wellendorff, Jess; Lundgaard, Keld T.; Jacobsen, Karsten W.
- The Journal of Chemical Physics, Vol. 140, Issue 14
M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics
journal, December 2011
- Peverati, Roberto; Truhlar, Donald G.
- The Journal of Physical Chemistry Letters, Vol. 3, Issue 1
High-Level Ab Initio Electronic Structure Calculations of Water Clusters (H 2 O) 16 and (H 2 O) 17 : A New Global Minimum for (H 2 O) 16
journal, October 2010
- Yoo, Soohaeng; Aprà, Edoardo; Zeng, Xiao Cheng
- The Journal of Physical Chemistry Letters, Vol. 1, Issue 20
Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods
journal, December 2011
- Řezáč, Jan; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 8, Issue 1
Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions
journal, October 2008
- Zhao, Yan; Truhlar, Donald G.
- Journal of Chemical Theory and Computation, Vol. 4, Issue 11
Variational, Self-Consistent Implementation of the Perdew–Zunger Self-Interaction Correction with Complex Optimal Orbitals
journal, November 2014
- Lehtola, Susi; Jónsson, Hannes
- Journal of Chemical Theory and Computation, Vol. 10, Issue 12
Use of the rVV10 Nonlocal Correlation Functional in the B97M-V Density Functional: Defining B97M-rV and Related Functionals
journal, December 2016
- Mardirossian, Narbe; Ruiz Pestana, Luis; Womack, James C.
- The Journal of Physical Chemistry Letters, Vol. 8, Issue 1
Long-range corrected hybrid meta-generalized-gradient approximations with dispersion corrections
journal, April 2012
- Lin, You-Sheng; Tsai, Chen-Wei; Li, Guan-De
- The Journal of Chemical Physics, Vol. 136, Issue 15
Hybrid Density Functional Methods Empirically Optimized for the Computation of 13 C and 1 H Chemical Shifts in Chloroform Solution
journal, May 2006
- Wiitala, Keith W.; Hoye, Thomas R.; Cramer, Christopher J.
- Journal of Chemical Theory and Computation, Vol. 2, Issue 4
Accuracy of Several Wave Function and Density Functional Theory Methods for Description of Noncovalent Interaction of Saturated and Unsaturated Hydrocarbon Dimers
journal, June 2012
- Granatier, Jaroslav; Pitoňák, Michal; Hobza, Pavel
- Journal of Chemical Theory and Computation, Vol. 8, Issue 7
Electrostatic Domination of the Effect of Electron Correlation in Intermolecular Interactions
journal, March 2014
- Thirman, Jonathan; Head-Gordon, Martin
- The Journal of Physical Chemistry Letters, Vol. 5, Issue 8
Investigation of Exchange Energy Density Functional Accuracy for Interacting Molecules
journal, August 2009
- Murray, Éamonn D.; Lee, Kyuho; Langreth, David C.
- Journal of Chemical Theory and Computation, Vol. 5, Issue 10
SCAN-based hybrid and double-hybrid density functionals from models without fitted parameters
journal, January 2016
- Hui, Kerwin; Chai, Jeng-Da
- The Journal of Chemical Physics, Vol. 144, Issue 4
Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions
journal, January 2006
- Zhao, Yan; Schultz, Nathan E.; Truhlar, Donald G.
- Journal of Chemical Theory and Computation, Vol. 2, Issue 2
Constrained Density Functional Theory
journal, November 2011
- Kaduk, Benjamin; Kowalczyk, Tim; Van Voorhis, Troy
- Chemical Reviews, Vol. 112, Issue 1
A multicenter numerical integration scheme for polyatomic molecules
journal, February 1988
- Becke, A. D.
- The Journal of Chemical Physics, Vol. 88, Issue 4
Conformational Equilibria in Butane-1,4-diol: A Benchmark of a Prototypical System with Strong Intramolecular H-bonds
journal, December 2013
- Kozuch, Sebastian; Bachrach, Steven M.; Martin, Jan M. L.
- The Journal of Physical Chemistry A, Vol. 118, Issue 1
Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits
journal, August 2005
- Perdew, John P.; Ruzsinszky, Adrienn; Tao, Jianmin
- The Journal of Chemical Physics, Vol. 123, Issue 6
ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy
journal, January 2014
- Mardirossian, Narbe; Head-Gordon, Martin
- Physical Chemistry Chemical Physics, Vol. 16, Issue 21
Exchange-correlation energy of a metallic surface: Wave-vector analysis
journal, March 1977
- Langreth, David C.; Perdew, John P.
- Physical Review B, Vol. 15, Issue 6
Screened-exchange density functionals with broad accuracy for chemistry and solid-state physics
journal, January 2012
- Peverati, Roberto; Truhlar, Donald G.
- Physical Chemistry Chemical Physics, Vol. 14, Issue 47
Influence of the exchange screening parameter on the performance of screened hybrid functionals
journal, December 2006
- Krukau, Aliaksandr V.; Vydrov, Oleg A.; Izmaylov, Artur F.
- The Journal of Chemical Physics, Vol. 125, Issue 22
Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections
journal, November 2012
- Lin, You-Sheng; Li, Guan-De; Mao, Shan-Ping
- Journal of Chemical Theory and Computation, Vol. 9, Issue 1
A new parametrization of exchange–correlation generalized gradient approximation functionals
journal, April 2001
- Boese, A. Daniel; Handy, Nicholas C.
- The Journal of Chemical Physics, Vol. 114, Issue 13
Accurate Semilocal Density Functional for Condensed-Matter Physics and Quantum Chemistry
journal, August 2016
- Tao, Jianmin; Mo, Yuxiang
- Physical Review Letters, Vol. 117, Issue 7
Local hybrid functionals
journal, January 2003
- Jaramillo, Juanita; Scuseria, Gustavo E.; Ernzerhof, Matthias
- The Journal of Chemical Physics, Vol. 118, Issue 3
Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory
journal, September 2012
- Klimeš, Jiří; Michaelides, Angelos
- The Journal of Chemical Physics, Vol. 137, Issue 12
Double-hybrid density functionals: Double-hybrid density functionals
journal, July 2014
- Goerigk, Lars; Grimme, Stefan
- Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 4, Issue 6
Characterizing and Understanding the Remarkably Slow Basis Set Convergence of Several Minnesota Density Functionals for Intermolecular Interaction Energies
journal, September 2013
- Mardirossian, Narbe; Head-Gordon, Martin
- Journal of Chemical Theory and Computation, Vol. 9, Issue 10
Heats of formation of platonic hydrocarbon cages by means of high-level thermochemical procedures
journal, June 2015
- Karton, Amir; Schreiner, Peter R.; Martin, Jan M. L.
- Journal of Computational Chemistry, Vol. 37, Issue 1
The calculation of atomic fields
journal, January 1927
- Thomas, L. H.
- Mathematical Proceedings of the Cambridge Philosophical Society, Vol. 23, Issue 5
An Assessment of Theoretical Methods for Nonbonded Interactions: Comparison to Complete Basis Set Limit Coupled-Cluster Potential Energy Curves for the Benzene Dimer, the Methane Dimer, Benzene−Methane, and Benzene−H 2 S †
journal, September 2009
- Sherrill, C. David; Takatani, Tait; Hohenstein, Edward G.
- The Journal of Physical Chemistry A, Vol. 113, Issue 38
Does the ionization potential condition employed in QTP functionals mitigate the self-interaction error?
journal, January 2017
- Ranasinghe, Duminda S.; Margraf, Johannes T.; Jin, Yifan
- The Journal of Chemical Physics, Vol. 146, Issue 3
Left-right correlation energy
journal, March 2001
- Handy, Nicholas C.; Cohen, Aron J.
- Molecular Physics, Vol. 99, Issue 5
Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions
journal, June 2005
- Zhao, Yan; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 109, Issue 25
Generalized gradient approximation model exchange holes for range-separated hybrids
journal, May 2008
- Henderson, Thomas M.; Janesko, Benjamin G.; Scuseria, Gustavo E.
- The Journal of Chemical Physics, Vol. 128, Issue 19
An accurate benchmark description of the interactions between carbon dioxide and polyheterocyclic aromatic compounds containing nitrogen
journal, January 2015
- Li, Sicheng; Smith, Daniel G. A.; Patkowski, Konrad
- Physical Chemistry Chemical Physics, Vol. 17, Issue 25
Development and assessment of new exchange-correlation functionals
journal, October 1998
- Hamprecht, Fred A.; Cohen, Aron J.; Tozer, David J.
- The Journal of Chemical Physics, Vol. 109, Issue 15
Magnetic-field density-functional theory
journal, October 1994
- Grayce, Christopher J.; Harris, Robert A.
- Physical Review A, Vol. 50, Issue 4
Performance of Density Functional Theory Procedures for the Calculation of Proton-Exchange Barriers: Unusual Behavior of M06-Type Functionals
journal, July 2014
- Chan, Bun; Gilbert, Andrew T. B.; Gill, Peter M. W.
- Journal of Chemical Theory and Computation, Vol. 10, Issue 9
Works referencing / citing this record:
Supramolecular Interactions of Fullerene C 60 with 1,3,5-Trifluoro-2,4,6-triiodobenzene: A Combined Theoretical and Experimental Study
journal, May 2018
- Zhang, Yu; Wang, Jian-Ge; Sun, Xiaotian
- ChemPlusChem, Vol. 83, Issue 5
The Selective N -Functionalization of Indoles via aza -Michael Addition in the Ligand Sphere of a Chiral Nickel(II) Complex: Asymmetric Synthesis of ( S )-1 H -Indole-Alanine Derivatives : The Selective N-Functionalization of Indoles via aza-Michael Addition in the Ligand Sphere of a Chiral Nickel(II) Complex: Asymmetric Synthesis of (S)
journal, May 2019
- Larionov, Vladimir A.; Savel'yeva, Tat'yana F.; Medvedev, Michael G.
- European Journal of Organic Chemistry, Vol. 2019, Issue 22
A Benchmark of Density Functional Approximations For Thermochemistry and Kinetics of Hydride Reductions of Cyclohexanones
journal, June 2019
- Deraet, Xavier; Woller, Tatiana; Van Lommel, Ruben
- ChemistryOpen, Vol. 8, Issue 6
Theoretical study of the photophysical processes of a styryl‐bodipy derivative eliciting an AND molecular logic gate response
journal, April 2019
- Tzeli, Demeter; Petsalakis, Ioannis D.; Theodorakopoulos, Giannoula
- International Journal of Quantum Chemistry, Vol. 119, Issue 16
Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning
journal, July 2019
- Smith, Justin S.; Nebgen, Benjamin T.; Zubatyuk, Roman
- Nature Communications, Vol. 10, Issue 1
Probing solvation and reactivity in ionized polycyclic aromatic hydrocarbon–water clusters with photoionization mass spectrometry and electronic structure calculations
journal, January 2019
- Xu, Bo; Stein, Tamar; Ablikim, Utuq
- Faraday Discussions, Vol. 217
Inner-shell X-ray absorption spectra of the cationic series NH y + ( y = 0–3)
journal, January 2019
- Bari, Sadia; Inhester, Ludger; Schubert, Kaja
- Physical Chemistry Chemical Physics, Vol. 21, Issue 30
Accurate quantum chemical energies for 133 000 organic molecules
journal, January 2019
- Narayanan, Badri; Redfern, Paul C.; Assary, Rajeev S.
- Chemical Science, Vol. 10, Issue 31
Gas phase formation of c-SiC 3 molecules in the circumstellar envelope of carbon stars
journal, July 2019
- Yang, Tao; Bertels, Luke; Dangi, Beni B.
- Proceedings of the National Academy of Sciences, Vol. 116, Issue 29
Structure and properties of some chiralanes and chirolanes
journal, February 2018
- Novak, Igor
- Molecular Physics, Vol. 116, Issue 12
Push it to the limit: comparing periodic and local approaches to density functional theory for intermolecular interactions
journal, June 2018
- Witte, Jonathon; Neaton, Jeffrey B.; Head-Gordon, Martin
- Molecular Physics, Vol. 117, Issue 9-12
Molecular dynamics of the intramolecular 1, 3-dipolar ene reaction of a nitrile oxide and an alkene: non-statistical behavior of a reaction involving a diradical intermediate
journal, June 2018
- Yu, Yanmin; Yang, Zhongyue; Houk, K. N.
- Molecular Physics, Vol. 117, Issue 9-12
Structures and binding energies for complexations of different spin states of Ni + and Ni 2+ to aromatic molecules
journal, June 2018
- Kerkeni, Boutheïna; Aquino, Adelia J. A.; Berman, Michael R.
- Molecular Physics, Vol. 117, Issue 9-12
Oxidative decomposition mechanisms of lithium peroxide clusters: an Ab Initio study
journal, June 2018
- Assary, Rajeev S.; Curtiss, Larry A.
- Molecular Physics, Vol. 117, Issue 9-12
Solvent effect on Al(III) hydrolysis constants from density functional theory
journal, June 2018
- Ridley, Moira K.; Lischka, Hans; Tunega, Daniel
- Molecular Physics, Vol. 117, Issue 9-12
Highly efficient perovskite solar cells by tuning electronic structures of thienothiophene-based as hole transport materials
journal, July 2019
- Han, Bingjie; Li, Zhuo; Li, Yuanzuo
- Molecular Physics
Constructing transformation paths for conformational changes in (MgF 2 ) n clusters using a stochastic procedure
journal, July 2019
- Mirdha, Rijaul Haque; Naskar, Pulak; Chaudhury, Pinaki
- Molecular Physics
Photoionization and photofragmentation of singly charged positive and negative endohedral fullerene ions
journal, June 2019
- Müller, A.; Martins, M.; Kilcoyne, A. L. D.
- Physical Review A, Vol. 99, Issue 6
Odd-even effect of the number of free valence electrons on the electronic structure properties of gold-thiolate clusters
text, January 2019
- Li, Yanle; Liu, Chunyan; Oliveira, Vytor
- Figshare
A Systematic Protocol for Benchmarking Guest-Host Interactions by First-Principles Computations: Capturing CO 2 in Clathrate Hydrates
journal, June 2018
- Arismendi-Arrieta, Daniel J.; Valdés, Álvaro; Prosmiti, Rita
- Chemistry - A European Journal, Vol. 24, Issue 37
Quantum Chemical Modeling of Pressure‐Induced Spin Crossover in Octahedral Metal‐Ligand Complexes
journal, October 2019
- Stauch, Tim; Chakraborty, Romit; Head‐Gordon, Martin
- ChemPhysChem, Vol. 20, Issue 21
Molecular Electrocatalysts for the Hydrogen Evolution Reaction: Input from Quantum Chemistry
journal, October 2019
- Barrozo, Alexandre; Orio, Maylis
- ChemSusChem, Vol. 12, Issue 22
Gaussian Process Regression Models for the Prediction of Hydrogen Bond Acceptor Strengths
journal, November 2018
- Bauer, Christoph A.; Schneider, Gisbert; Göller, Andreas H.
- Molecular Informatics, Vol. 38, Issue 4
Fully numerical Hartree‐Fock and density functional calculations. I. Atoms
journal, April 2019
- Lehtola, Susi
- International Journal of Quantum Chemistry, Vol. 119, Issue 19
Making machine learning a useful tool in the accelerated discovery of transition metal complexes
journal, July 2019
- Kulik, Heather J.
- WIREs Computational Molecular Science, Vol. 10, Issue 1
Template effects on the pressure-dependent behavior of chabazite-type fluoroaluminophosphates: a computational approach
journal, November 2018
- Fischer, Michael
- Physics and Chemistry of Minerals, Vol. 46, Issue 4
Electronic structure and second-order nonlinear optical property of chiral peropyrenes
journal, July 2019
- Gong, Lijing; Liu, Chunyu; Du, Xin
- Journal of Molecular Modeling, Vol. 25, Issue 8
A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions
journal, January 2017
- Goerigk, Lars; Hansen, Andreas; Bauer, Christoph
- Physical Chemistry Chemical Physics, Vol. 19, Issue 48
Barriometry – an enhanced database of accurate barrier heights for gas-phase reactions
journal, January 2018
- Chan, Bun; Simmie, John M.
- Physical Chemistry Chemical Physics, Vol. 20, Issue 16
How accurate are static polarizability predictions from density functional theory? An assessment over 132 species at equilibrium geometry
journal, January 2018
- Hait, Diptarka; Head-Gordon, Martin
- Physical Chemistry Chemical Physics, Vol. 20, Issue 30
Semi-empirical or non-empirical double-hybrid density functionals: which are more robust?
journal, January 2018
- Mehta, Nisha; Casanova-Páez, Marcos; Goerigk, Lars
- Physical Chemistry Chemical Physics, Vol. 20, Issue 36
‘Diet GMTKN55’ offers accelerated benchmarking through a representative subset approach
journal, January 2018
- Gould, Tim
- Physical Chemistry Chemical Physics, Vol. 20, Issue 44
A first-principles roadmap and limits to design efficient supercapacitor electrode materials
journal, January 2019
- Ali, Basant A.; Allam, Nageh K.
- Physical Chemistry Chemical Physics, Vol. 21, Issue 32
How accurate are approximate quantum chemical methods at modelling solute–solvent interactions in solvated clusters?
journal, January 2020
- Chen, Junbo; Chan, Bun; Shao, Yihan
- Physical Chemistry Chemical Physics, Vol. 22, Issue 7
Valence electronic structure of [EMIM][B(CN) 4 ]: ion-pair vs. bulk description
journal, January 2019
- Kuusik, I.; Berholts, M.; Kruusma, J.
- RSC Advances, Vol. 9, Issue 57
Electron density learning of non-covalent systems
journal, January 2019
- Fabrizio, Alberto; Grisafi, Andrea; Meyer, Benjamin
- Chemical Science, Vol. 10, Issue 41
Sulfamides direct radical-mediated chlorination of aliphatic C–H bonds
journal, January 2020
- Short, Melanie A.; Shehata, Mina F.; Sanders, Matthew A.
- Chemical Science, Vol. 11, Issue 1
Communication: xDH double hybrid functionals can be qualitatively incorrect for non-equilibrium geometries: Dipole moment inversion and barriers to radical-radical association using XYG3 and XYGJ-OS
journal, May 2018
- Hait, Diptarka; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 148, Issue 17
The S66 Non-Covalent Interactions Benchmark Reconsidered Using Explicitly Correlated Methods Near the Basis Set Limit
journal, January 2018
- Kesharwani, Manoj K.; Karton, Amir; Sylvetsky, Nitai
- Australian Journal of Chemistry, Vol. 71, Issue 4
EPR parameters of L - α -alanine radicals in aqueous solution: a first-principles study
journal, May 2018
- Janbazi, Mehdi; T. Azar, Yavar; Ziaie, Farhood
- Molecular Physics, Vol. 116, Issue 14
Quantum chemical study on gas phase decomposition of ferulic acid
journal, April 2018
- Verma, Anand Mohan; Agrawal, Kushagra; Kawale, Harshal D.
- Molecular Physics, Vol. 116, Issue 14
Theoretical study of the mechanism of 2,5-diketopiperazine formation during pyrolysis of proline
journal, March 2019
- Cervantes, Cristian; Mora, José R.; Rincón, Luis
- Molecular Physics, Vol. 118, Issue 2
Theoretical insights into elaborating and regulating excited state dynamics for the novel 6-cyano-2-(2′-hydroxyphenyl)imidazo[1,2a]pyridine system in polar and nonpolar solvents
journal, September 2019
- Xu, Lei; Zhang, Qiaoli; Zhang, Tianjie
- Molecular Physics, Vol. 118, Issue 8
Machine learning prediction of accurate atomization energies of organic molecules from low-fidelity quantum chemical calculations
journal, August 2019
- Ward, Logan; Blaiszik, Ben; Foster, Ian
- MRS Communications, Vol. 9, Issue 3
Intermolecular Interactions in Molecular Organic Crystals upon Relaxation of Lattice Parameters
journal, December 2019
- Stein, Matthias; Heimsaat, Madalen
- Crystals, Vol. 9, Issue 12
Figures / Tables found in this record: