
Geometric Energy Derivatives at the Complete Basis Set Limit: Application to the Equilibrium Structure and Molecular Force Field of Formaldehyde
Abstract
Here, geometric energy derivatives which rely on core-corrected focal-point energies extrapolated to the complete basis set (CBS) limit of coupled cluster theory with iterative and noniterative quadruple excitations, CCSDTQ and CCSDT(Q), are used as elements of molecular gradients and, in the case of CCSDT(Q), expansion coefficients of an anharmonic force field. These gradients are used to determine the CCSDTQ/CBS and CCSDT(Q)/CBS equilibrium structure of the S0 ground state of H2CO where excellent agreement is observed with previous work and experimentally derived results. A fourth-order expansion about this CCSDT(Q)/CBS reference geometry using the same level of theory produces an exceptional level of agreement to spectroscopically observed vibrational band origins with a MAE of 0.57 cm–1. Second-order vibrational perturbation theory (VPT2) and variational discrete variable representation (DVR) results are contrasted and discussed. Vibration–rotation, anharmonicity, and centrifugal distortion constants from the VPT2 analysis are reported and compared to previous work. Additionally, an initial application of a sum-over-states fourth-order vibrational perturbation theory (VPT4) formalism is employed herein, utilizing quintic and sextic derivatives obtained with a recursive algorithmic approach for response theory.
- Authors:
-
 [1];
[1];
[4];
[1]
[1];
[1];
[4];
[1]
- Univ. of Georgia, Athens, GA (United States)
- Univ. of Texas at Austin, Austin, TX (United States)
- Univ. of Tromso - The Arctic Univ. of Norway, Tromso (Norway)
- Univ. of Florida, Gainesville, FL (United States)
- Publication Date:
- Research Org.:
- Univ. of Georgia, Athens, GA (United States)
- Sponsoring Org.:
- USDOE Office of Science (SC), Basic Energy Sciences (BES)
- OSTI Identifier:
- 1468297
- Grant/Contract Number:
- SC0018412
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Journal of Chemical Theory and Computation
- Additional Journal Information:
- Journal Volume: 14; Journal Issue: 3; Journal ID: ISSN 1549-9618
- Publisher:
- American Chemical Society
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 71 CLASSICAL AND QUANTUM MECHANICS, GENERAL PHYSICS
Citation Formats
Morgan, W. James, Matthews, Devin A., Ringholm, Magnus, Agarwal, Jay, Gong, Justin Z., Ruud, Kenneth, Allen, Wesley D., Stanton, John F., and Schaefer, III, Henry F. Geometric Energy Derivatives at the Complete Basis Set Limit: Application to the Equilibrium Structure and Molecular Force Field of Formaldehyde. United States: N. p., 2018.
Web. doi:10.1021/acs.jctc.7b01138.
Morgan, W. James, Matthews, Devin A., Ringholm, Magnus, Agarwal, Jay, Gong, Justin Z., Ruud, Kenneth, Allen, Wesley D., Stanton, John F., & Schaefer, III, Henry F. Geometric Energy Derivatives at the Complete Basis Set Limit: Application to the Equilibrium Structure and Molecular Force Field of Formaldehyde. United States. https://doi.org/10.1021/acs.jctc.7b01138
Morgan, W. James, Matthews, Devin A., Ringholm, Magnus, Agarwal, Jay, Gong, Justin Z., Ruud, Kenneth, Allen, Wesley D., Stanton, John F., and Schaefer, III, Henry F. Fri .
"Geometric Energy Derivatives at the Complete Basis Set Limit: Application to the Equilibrium Structure and Molecular Force Field of Formaldehyde". United States. https://doi.org/10.1021/acs.jctc.7b01138. https://www.osti.gov/servlets/purl/1468297.
@article{osti_1468297,
title = {Geometric Energy Derivatives at the Complete Basis Set Limit: Application to the Equilibrium Structure and Molecular Force Field of Formaldehyde},
author = {Morgan, W. James and Matthews, Devin A. and Ringholm, Magnus and Agarwal, Jay and Gong, Justin Z. and Ruud, Kenneth and Allen, Wesley D. and Stanton, John F. and Schaefer, III, Henry F.},
abstractNote = {Here, geometric energy derivatives which rely on core-corrected focal-point energies extrapolated to the complete basis set (CBS) limit of coupled cluster theory with iterative and noniterative quadruple excitations, CCSDTQ and CCSDT(Q), are used as elements of molecular gradients and, in the case of CCSDT(Q), expansion coefficients of an anharmonic force field. These gradients are used to determine the CCSDTQ/CBS and CCSDT(Q)/CBS equilibrium structure of the S0 ground state of H2CO where excellent agreement is observed with previous work and experimentally derived results. A fourth-order expansion about this CCSDT(Q)/CBS reference geometry using the same level of theory produces an exceptional level of agreement to spectroscopically observed vibrational band origins with a MAE of 0.57 cm–1. Second-order vibrational perturbation theory (VPT2) and variational discrete variable representation (DVR) results are contrasted and discussed. Vibration–rotation, anharmonicity, and centrifugal distortion constants from the VPT2 analysis are reported and compared to previous work. Additionally, an initial application of a sum-over-states fourth-order vibrational perturbation theory (VPT4) formalism is employed herein, utilizing quintic and sextic derivatives obtained with a recursive algorithmic approach for response theory.},
doi = {10.1021/acs.jctc.7b01138},
journal = {Journal of Chemical Theory and Computation},
number = 3,
volume = 14,
place = {United States},
year = {Fri Feb 23 00:00:00 EST 2018},
month = {Fri Feb 23 00:00:00 EST 2018}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
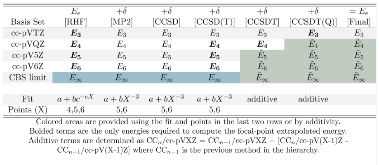 Table 1: Example Focal Point Table of CCSDT(Q)/CBS Energies
Table 1: Example Focal Point Table of CCSDT(Q)/CBS Energies
Works referenced in this record:
An accurate quartic force field for formaldehyde
journal, January 1996
- Burleigh, Darin C.; McCoy, Anne B.; Sibert, Edwin L.
- The Journal of Chemical Physics, Vol. 104, Issue 2
The vibrations of formaldehyde
journal, July 1995
- Carter, Stuart; Pinnavaia, Nadja; Handy, Nicholas C.
- Chemical Physics Letters, Vol. 240, Issue 5-6
The Geometry of Formaldehyde
journal, September 1996
- Carter, Stuart; Handy, Nicholas C.
- Journal of Molecular Spectroscopy, Vol. 179, Issue 1
A new “spectroscopic” potential energy surface for formaldehyde in its ground electronic state
journal, June 2011
- Yachmenev, Andrey; Yurchenko, Sergei N.; Jensen, Per
- The Journal of Chemical Physics, Vol. 134, Issue 24
High-Accuracy ab Initio Rotation-Vibration Transitions for Water
journal, January 2003
- Polyansky, O. L.
- Science, Vol. 299, Issue 5606
On equilibrium structures of the water molecule
journal, June 2005
- Császár, Attila G.; Czakó, Gábor; Furtenbacher, Tibor
- The Journal of Chemical Physics, Vol. 122, Issue 21
CVRQD ab initio ground-state adiabatic potential energy surfaces for the water molecule
journal, November 2006
- Barletta, Paolo; Shirin, Sergei V.; Zobov, Nikolai F.
- The Journal of Chemical Physics, Vol. 125, Issue 20
Performance of W4 theory for spectroscopic constants and electrical properties of small molecules
journal, October 2010
- Karton, Amir; Martin, Jan M. L.
- The Journal of Chemical Physics, Vol. 133, Issue 14
A global potential energy surface and dipole moment surface for silane
journal, December 2015
- Owens, Alec; Yurchenko, Sergei N.; Yachmenev, Andrey
- The Journal of Chemical Physics, Vol. 143, Issue 24
Calculation of rotation-vibration energy levels of the ammonia molecule based on an ab initio potential energy surface
journal, September 2016
- Polyansky, Oleg L.; Ovsyannikov, Roman I.; Kyuberis, Aleksandra A.
- Journal of Molecular Spectroscopy, Vol. 327
Full-Dimensional Potential Energy and Dipole Moment Surfaces of GeH 4 Molecule and Accurate First-Principle Rotationally Resolved Intensity Predictions in the Infrared
journal, November 2016
- Nikitin, A. V.; Rey, M.; Rodina, A.
- The Journal of Physical Chemistry A, Vol. 120, Issue 45
First fully ab initio potential energy surface of methane with a spectroscopic accuracy
journal, September 2016
- Nikitin, A. V.; Rey, M.; Tyuterev, Vl. G.
- The Journal of Chemical Physics, Vol. 145, Issue 11
In pursuit of the ab initio limit for conformational energy prototypes
journal, June 1998
- Császár, Attila G.; Allen, Wesley D.; Schaefer, Henry F.
- The Journal of Chemical Physics, Vol. 108, Issue 23
Characterization of the X ̃ 1 A ’ state of isocyanic acid
journal, January 1993
- East, Allan L. L.; Johnson, Christopher S.; Allen, Wesley D.
- The Journal of Chemical Physics, Vol. 98, Issue 2
The heat of formation of NCO
journal, September 1993
- East, Allan L. L.; Allen, Wesley D.
- The Journal of Chemical Physics, Vol. 99, Issue 6
Revisitation of Nonorthogonal Spin Adaptation in Coupled Cluster Theory
journal, May 2013
- Matthews, Devin A.; Gauss, Jürgen; Stanton, John F.
- Journal of Chemical Theory and Computation, Vol. 9, Issue 6
The full CCSDT model for molecular electronic structure
journal, June 1987
- Noga, Jozef; Bartlett, Rodney J.
- The Journal of Chemical Physics, Vol. 86, Issue 12
Erratum: The full CCSDT model for molecular electronic structure [J. Chem. Phys. 8 6 , 7041 (1987)]
journal, September 1988
- Noga, J.; Bartlett, R. J.
- The Journal of Chemical Physics, Vol. 89, Issue 5
Coupled‐cluster method truncated at quadruples
journal, November 1991
- Oliphant, Nevin; Adamowicz, Ludwik
- The Journal of Chemical Physics, Vol. 95, Issue 9
The coupled‐cluster single, double, triple, and quadruple excitation method
journal, September 1992
- Kucharski, Stanislaw A.; Bartlett, Rodney J.
- The Journal of Chemical Physics, Vol. 97, Issue 6
Coupled-cluster methods including noniterative corrections for quadruple excitations
journal, August 2005
- Bomble, Yannick J.; Stanton, John F.; Kállay, Mihály
- The Journal of Chemical Physics, Vol. 123, Issue 5
Non-orthogonal spin-adaptation of coupled cluster methods: A new implementation of methods including quadruple excitations
journal, February 2015
- Matthews, Devin A.; Stanton, John F.
- The Journal of Chemical Physics, Vol. 142, Issue 6
Accelerating the convergence of higher-order coupled cluster methods
journal, November 2015
- Matthews, Devin A.; Stanton, John F.
- The Journal of Chemical Physics, Vol. 143, Issue 20
Stimulated emission spectroscopy: A complete set of vibrational constants for X ̃ 1 A 1 formaldehyde
journal, June 1984
- Reisner, David E.; Field, Robert W.; Kinsey, James L.
- The Journal of Chemical Physics, Vol. 80, Issue 12
Stark level‐crossing spectroscopy of S 0 formaldehyde eigenstates at the dissociation threshold
journal, March 1990
- Polik, William F.; Guyer, Dean R.; Moore, C. Bradley
- The Journal of Chemical Physics, Vol. 92, Issue 6
Eigenstate‐resolved unimolecular reaction dynamics: Ergodic character of S 0 formaldehyde at the dissociation threshold
journal, March 1990
- Polik, William F.; Guyer, Dean R.; Miller, William H.
- The Journal of Chemical Physics, Vol. 92, Issue 6
Pure vibrational spectroscopy of S 0 formaldehyde by dispersed fluorescence
journal, January 1996
- Bouwens, Rychard J.; Hammerschmidt, Jon A.; Grzeskowiak, Martha M.
- The Journal of Chemical Physics, Vol. 104, Issue 2
High accuracy measurements on the ground state rotational spectrum of formaldehyde (H2CO) up to 2 THzElectronic supplementary information (ESI) available: Newly measured pure rotational transitions. See http://www.rsc.org/suppdata/cp/b3/b301657a/
journal, March 2003
- Brünken, S.; Müller, H. S. P.; Lewen, F.
- Physical Chemistry Chemical Physics, Vol. 5, Issue 8
FTFIR-spectrum of the ground state of D2CO
journal, November 2004
- Lohilahti, Jarmo; Horneman, Veli-Matti
- Journal of Molecular Spectroscopy, Vol. 228, Issue 1
The HITRAN2016 molecular spectroscopic database
journal, December 2017
- Gordon, I. E.; Rothman, L. S.; Hill, C.
- Journal of Quantitative Spectroscopy and Radiative Transfer, Vol. 203
Spectroscopy of the formaldehyde isotopomer H213CO in the microwave to terahertz region
journal, January 2000
- Müller, H. S. P.; Gendriesch, R.; Margulès, L.
- Physical Chemistry Chemical Physics, Vol. 2, Issue 15
The accuracy of rotational constants predicted by high-level quantum-chemical calculations. I. molecules containing first-row atoms
journal, May 2008
- Puzzarini, Cristina; Heckert, Miriam; Gauss, Jürgen
- The Journal of Chemical Physics, Vol. 128, Issue 19
The ab initio limit quartic force field of BH 3
journal, August 2005
- Schuurman, Michael S.; Allen, Wesley D.; Schaefer, Henry F.
- Journal of Computational Chemistry, Vol. 26, Issue 11
A procedure for computing accurate ab initio quartic force fields: Application to HO2+ and H2O
journal, July 2008
- Huang, Xinchuan; Lee, Timothy J.
- The Journal of Chemical Physics, Vol. 129, Issue 4
Accurate ab initio quartic force fields for NH[sub 2]−] and CCH[sup −] and rovibrational spectroscopic constants for their isotopologs
journal, January 2009
- Huang, Xinchuan; Lee, Timothy J.
- The Journal of Chemical Physics, Vol. 131, Issue 10
The trans -HOCO radical: Quartic force fields, vibrational frequencies, and spectroscopic constants
journal, October 2011
- Fortenberry, Ryan C.; Huang, Xinchuan; Francisco, Joseph S.
- The Journal of Chemical Physics, Vol. 135, Issue 13
Quartic force fields for excited electronic states: Rovibronic reference data for the 1 2 A ′ 2 A ″
journal, January 2015
- Morgan, W. James; Fortenberry, Ryan C.
- Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 135
A general, recursive, and open-ended response code
journal, February 2014
- Ringholm, Magnus; Jonsson, Dan; Ruud, Kenneth
- Journal of Computational Chemistry, Vol. 35, Issue 8
Benchmark calculations with correlated molecular wave functions. VII. Binding energy and structure of the HF dimer
journal, February 1995
- Peterson, Kirk A.; Dunning, Thom H.
- The Journal of Chemical Physics, Vol. 102, Issue 5
Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions
journal, May 1992
- Kendall, Rick A.; Dunning, Thom H.; Harrison, Robert J.
- The Journal of Chemical Physics, Vol. 96, Issue 9
Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited
journal, June 2001
- Dunning, Thom H.; Peterson, Kirk A.; Wilson, Angela K.
- The Journal of Chemical Physics, Vol. 114, Issue 21
The use of systematic sequences of wave functions for estimating the complete basis set, full configuration interaction limit in water
journal, May 1993
- Feller, David
- The Journal of Chemical Physics, Vol. 98, Issue 9
Basis-set convergence of correlated calculations on water
journal, June 1997
- Helgaker, Trygve; Klopper, Wim; Koch, Henrik
- The Journal of Chemical Physics, Vol. 106, Issue 23
Importance of Angular Correlations between Atomic Electrons
journal, May 1962
- Schwartz, Charles
- Physical Review, Vol. 126, Issue 3
Rates of convergence of the partial‐wave expansions of atomic correlation energies
journal, March 1992
- Kutzelnigg, Werner; Morgan, John D.
- The Journal of Chemical Physics, Vol. 96, Issue 6
Erratum: Rates of convergence of the partial‐wave expansions of atomic correlation energies [J. Chem. Phys. 96 , 4484 (1992)]
journal, December 1992
- Kutzelnigg, W.; Morgan, J. D.
- The Journal of Chemical Physics, Vol. 97, Issue 11
On the effectiveness of CCSD(T) complete basis set extrapolations for atomization energies
journal, July 2011
- Feller, David; Peterson, Kirk A.; Grant Hill, J.
- The Journal of Chemical Physics, Vol. 135, Issue 4
The atomization energy and proton affinity of NH3. An ab initio calibration study
journal, August 1996
- Martin, Jan M. L.; Lee, Timothy J.
- Chemical Physics Letters, Vol. 258, Issue 1-2
The extrapolation of one-electron basis sets in electronic structure calculations: How it should work and how it can be made to work
journal, January 2005
- Schwenke, David W.
- The Journal of Chemical Physics, Vol. 122, Issue 1
HEAT: High accuracy extrapolated ab initio thermochemistry
journal, December 2004
- Tajti, Attila; Szalay, Péter G.; Császár, Attila G.
- The Journal of Chemical Physics, Vol. 121, Issue 23
High-accuracy extrapolated ab initio thermochemistry. II. Minor improvements to the protocol and a vital simplification
journal, August 2006
- Bomble, Yannick J.; Vázquez, Juana; Kállay, Mihály
- The Journal of Chemical Physics, Vol. 125, Issue 6
High-accuracy extrapolated ab initio thermochemistry. III. Additional improvements and overview
journal, March 2008
- Harding, Michael E.; Vázquez, Juana; Ruscic, Branko
- The Journal of Chemical Physics, Vol. 128, Issue 11
Psi4: an open-source ab initio electronic structure program : Psi4: an electronic structure program
journal, October 2011
- Turney, Justin M.; Simmonett, Andrew C.; Parrish, Robert M.
- Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 2, Issue 4
Approximate relativistic corrections to atomic radial wave functions*
journal, January 1976
- Cowan, Robert D.; Griffin, Donald C.
- Journal of the Optical Society of America, Vol. 66, Issue 10
Analytic second derivatives in high-order many-body perturbation and coupled-cluster theories: Computational considerations and applications
journal, January 2000
- Stanton, John F.; Gauss, Jurgen
- International Reviews in Physical Chemistry, Vol. 19, Issue 1
Analytic evaluation of the dipole Hessian matrix in coupled-cluster theory
journal, October 2013
- Jagau, Thomas-C.; Gauss, Jürgen; Ruud, Kenneth
- The Journal of Chemical Physics, Vol. 139, Issue 15
Molecular equilibrium geometries based on coupled-cluster calculations including quadruple excitations
journal, August 2005
- Heckert, Miriam; KÁllay ‡, MihÁly; Gauss †, Jürgen
- Molecular Physics, Vol. 103, Issue 15-16
Basis-set extrapolation techniques for the accurate calculation of molecular equilibrium geometries using coupled-cluster theory
journal, July 2006
- Heckert, Miriam; Kállay, Mihály; Tew, David P.
- The Journal of Chemical Physics, Vol. 125, Issue 4
An Accurate ab Initio Quartic Force Field for Formaldehyde and Its Isotopomers
journal, July 1993
- Martin, J. M. L.; Lee, T. J.; Taylor, P. R.
- Journal of Molecular Spectroscopy, Vol. 160, Issue 1
Calibration-quality adiabatic potential energy surfaces for H3+ and its isotopologues
journal, May 2012
- Pavanello, Michele; Adamowicz, Ludwik; Alijah, Alexander
- The Journal of Chemical Physics, Vol. 136, Issue 18
Spectroscopy of H 3 + based on a new high-accuracy global potential energy surface
journal, November 2012
- Polyansky, Oleg L.; Alijah, Alexander; Zobov, Nikolai F.
- Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, Vol. 370, Issue 1978
An accurate quartic force field and vibrational frequencies for HNO and DNO
journal, October 1994
- Dateo, Christopher E.; Lee, Timothy J.; Schwenke, David W.
- The Journal of Chemical Physics, Vol. 101, Issue 7
General derivative relations for anharmonic force fields
journal, December 1996
- Allen, Wesley D.; CsÁSzÁR, Attila G.; Szalay, Viktor
- Molecular Physics, Vol. 89, Issue 5
A density matrix-based quasienergy formulation of the Kohn–Sham density functional response theory using perturbation- and time-dependent basis sets
journal, December 2008
- Thorvaldsen, Andreas J.; Ruud, Kenneth; Kristensen, Kasper
- The Journal of Chemical Physics, Vol. 129, Issue 21
The Dalton quantum chemistry program system: The Dalton program
journal, September 2013
- Aidas, Kestutis; Angeli, Celestino; Bak, Keld L.
- Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 4, Issue 3
GEN1INT: A unified procedure for the evaluation of one-electron integrals over Gaussian basis functions and their geometric derivatives
journal, August 2010
- Gao, Bin; Thorvaldsen, Andreas J.; Ruud, Kenneth
- International Journal of Quantum Chemistry, Vol. 111, Issue 4
A unified scheme for the calculation of differentiated and undifferentiated molecular integrals over solid-harmonic Gaussians
journal, January 2007
- Reine, Simen; Tellgren, Erik; Helgaker, Trygve
- Physical Chemistry Chemical Physics, Vol. 9, Issue 34
Linear response calculations for large scale multiconfiguration self‐consistent field wave functions
journal, September 1988
- Jo/rgensen, Poul; Jensen, Hans Jo/rgen Aagaard; Olsen, Jeppe
- The Journal of Chemical Physics, Vol. 89, Issue 6
Efficient elimination of response parameters in molecular property calculations for variational and nonvariational energies
journal, December 2008
- Kristensen, Kasper; Jørgensen, Poul; Thorvaldsen, Andreas J.
- The Journal of Chemical Physics, Vol. 129, Issue 21
Higher Order Rotation‐Vibration Energies of Polyatomic Molecules. IV
journal, September 1958
- Amat, Gilbert; Nielsen, Harald H.
- The Journal of Chemical Physics, Vol. 29, Issue 3
Theoretical studies of vibrationally excited polyatomic molecules using canonical Van Vleck perturbation theory
journal, April 1988
- Sibert, Edwin L.
- The Journal of Chemical Physics, Vol. 88, Issue 7
Numerical-Analytic Implementation of the Higher-Order Canonical Van Vleck Perturbation Theory for the Interpretation of Medium-Sized Molecule Vibrational Spectra
journal, March 2012
- Krasnoshchekov, Sergey V.; Isayeva, Elena V.; Stepanov, Nikolay F.
- The Journal of Physical Chemistry A, Vol. 116, Issue 14
The prediction of molecular equilibrium structures by the standard electronic wave functions
journal, April 1997
- Helgaker, Trygve; Gauss, Jürgen; Jo/rgensen, Poul
- The Journal of Chemical Physics, Vol. 106, Issue 15
The CO molecule: the role of basis set and correlation treatment in the calculation of molecular properties
journal, July 1997
- Peterson, Kirk A.; Dunning, Thom H.
- Journal of Molecular Structure: THEOCHEM, Vol. 400
Molecular Geometries at Sixth Order Møller−Plesset Perturbation Theory. At What Order Does MP Theory Give Exact Geometries?
journal, August 2000
- He, Yuan; Cremer, Dieter
- The Journal of Physical Chemistry A, Vol. 104, Issue 32
The accurate determination of molecular equilibrium structures
journal, April 2001
- Bak, Keld L.; Gauss, Jürgen; Jørgensen, Poul
- The Journal of Chemical Physics, Vol. 114, Issue 15
Exact geometries from quantum chemical calculations
journal, June 2001
- Cremer, Dieter; Kraka, Elfi; He, Yuan
- Journal of Molecular Structure, Vol. 567-568
A priori calculation of molecular properties to chemical accuracy
journal, August 2004
- Helgaker, Trygve; Ruden, Torgeir A.; Jørgensen, Poul
- Journal of Physical Organic Chemistry, Vol. 17, Issue 11
CCSDT calculations of molecular equilibrium geometries
journal, August 1997
- Halkier, Asger; Jørgensen, Poul; Gauss, Jürgen
- Chemical Physics Letters, Vol. 274, Issue 1-3
Communication: The performance of non-iterative coupled cluster quadruples models
journal, July 2015
- Eriksen, Janus J.; Matthews, Devin A.; Jørgensen, Poul
- The Journal of Chemical Physics, Vol. 143, Issue 4
Experimental, semi-experimental and ab initio equilibrium structures
journal, December 2007
- Demaison, J.
- Molecular Physics, Vol. 105, Issue 23-24
Magnetic interactions in molecules and an analysis of molecular electronic charge distribution from magnetic parameters
journal, December 1974
- Flygare, W. H.
- Chemical Reviews, Vol. 74, Issue 6
The Molecular Zeeman Effect of Imines. I. Methanimine, its Molecular g-Tensor, its Magnetic Susceptibility Anisotropies, its Molecular Electric Quadrupole Moment, its Electric Field Gradient at the Nitrogen Nucleus, and its Nitrogen Spin-Rotation Coupling
journal, November 1989
- Krause, H.; Sutter, D. H.
- Zeitschrift für Naturforschung A, Vol. 44, Issue 11
Submillimeterwave spectrum of CH2PH and equilibrium structures of CH2PH and CH2NH
journal, August 2006
- Margulès, L.; Demaison, J.; Sreeja, P. B.
- Journal of Molecular Spectroscopy, Vol. 238, Issue 2
Basis set convergence for geometry and harmonic frequencies. Are h functions enough?
journal, August 1994
- Martin, Jan M. L.; Taylor, Peter R.
- Chemical Physics Letters, Vol. 225, Issue 4-6
On the effect of core correlation on the geometry and harmonic frequencies of small polyatomic molecules
journal, August 1995
- Martin, Jan M. L.
- Chemical Physics Letters, Vol. 242, Issue 3
Basis Set Limit CCSD(T) Harmonic Vibrational Frequencies †
journal, November 2007
- Tew, David P.; Klopper, Wim; Heckert, Miriam
- The Journal of Physical Chemistry A, Vol. 111, Issue 44
Coupled-cluster connected quadruples and quintuples corrections to the harmonic vibrational frequencies and equilibrium bond distances of HF, N2, F2, and CO
journal, September 2004
- Ruden, Torgeir A.; Helgaker, Trygve; Jørgensen, Poul
- The Journal of Chemical Physics, Vol. 121, Issue 12
Factors Contributing to the Accuracy of Harmonic Force Field Calculations for Water
journal, May 2007
- Cortez, Michael H.; Brinkmann, Nicole R.; Polik, William F.
- Journal of Chemical Theory and Computation, Vol. 3, Issue 4
Analytic second derivatives for general coupled-cluster and configuration-interaction models
journal, April 2004
- Kállay, Mihály; Gauss, Jürgen
- The Journal of Chemical Physics, Vol. 120, Issue 15
The geometry, vibrational frequencies, and total atomization energy of ethylene. A calibration study
journal, January 1996
- Martin, Jan M. L.; Taylor, Peter R.
- Chemical Physics Letters, Vol. 248, Issue 5-6
Ab Initio Calibration Study of the Heat of Formation, Geometry, and Anharmonic Force Field of Fluoroacetylene
journal, March 1998
- Persson, B. Joakim; Taylor, Peter R.; Martin, Jan M. L.
- The Journal of Physical Chemistry A, Vol. 102, Issue 14
High level coupled cluster determination of the structure, frequencies, and heat of formation of water
journal, October 2009
- Feller, David; Peterson, Kirk A.
- The Journal of Chemical Physics, Vol. 131, Issue 15
Anharmonic vibrational analysis of water with traditional and explicitly correlated coupled cluster methods
journal, August 2010
- Kahn, Kalju; Kirtman, Bernard; Noga, Jozef
- The Journal of Chemical Physics, Vol. 133, Issue 7
A systematic study of molecular vibrational anharmonicity and vibration—rotation interaction by self-consistent-field higher-derivative methods. Asymmetric top molecules
journal, July 1988
- Clabo, D. Allen; Allen, Wesley D.; Remington, Richard B.
- Chemical Physics, Vol. 123, Issue 2
The Vibration-Rotation Energies of Polyatomic Molecules Part II. Accidental Degeneracies
journal, October 1945
- Nielsen, Harald H.
- Physical Review, Vol. 68, Issue 7-8
The anharmonic force field of ethylene, C 2 H 4 , by means of accurate ab initio calculations
journal, August 1995
- Martin, Jan M. L.; Lee, Timothy J.; Taylor, Peter R.
- The Journal of Chemical Physics, Vol. 103, Issue 7
Calculated stretching overtone levels and Darling–Dennison resonances in water: a triumph of simple theoretical approaches
journal, October 2007
- Matthews, Devin A.; Vázquez, Juana; Stanton, John F.
- Molecular Physics, Vol. 105, Issue 19-22
Quantitative analysis of Fermi resonances by harmonic derivatives of perturbation theory corrections
journal, October 2009
- Matthews, Devin; Stanton, John
- Molecular Physics, Vol. 107, Issue 3
Beyond the x - K relations : Calculations of 1–1 and 2–2 resonance constants with application to HCN and DCN
journal, April 1989
- Lehmann, Kevin K.
- Molecular Physics, Vol. 66, Issue 6
Accurate ab initio quartic force field for trans-HNNH and treatment of resonance polyads
journal, July 1997
- Martin, Jan M. L.; Taylor, Peter R.
- Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 53, Issue 8
VPT2+K spectroscopic constants and matrix elements of the transformed vibrational Hamiltonian of a polyatomic molecule with resonances using Van Vleck perturbation theory
journal, June 2013
- Rosnik, Andreana M.; Polik, William F.
- Molecular Physics, Vol. 112, Issue 2
Calculation of fundamental frequencies for small polyatomic molecules: a comparison between correlation consistent and atomic natural orbital basis sets
journal, May 2013
- McCaslin, Laura; Stanton, John
- Molecular Physics, Vol. 111, Issue 9-11
New analysis of the ν2 band of formaldehyde : Line positions for the ν2, ν3, ν4 and ν6 interacting bands
journal, October 2007
- Tchana, F. Kwabia; Perrin, A.; Lacome, N.
- Journal of Molecular Spectroscopy, Vol. 245, Issue 2
New analysis of the 2ν4, ν4+ν6, 2ν6, ν3+ν4, ν3+ν6, ν1, ν5, ν2+ν4, 2ν3, ν2+ν6 and ν2+ν3 bands of formaldehyde H212C16O: Line positions and intensities in the 3.5μm spectral region
journal, January 2006
- Perrin, A.; Valentin, A.; Daumont, L.
- Journal of Molecular Structure, Vol. 780-781
High resolution spectroscopy of H 2 12 C 16 O in the 1.9 to 2.56 µm spectral range
journal, June 2006
- Flaud, J. -M.; Lafferty, W. J.; Sams, R. L.
- Molecular Physics, Vol. 104, Issue 12
Efficient calculation of potential energy surfaces for the generation of vibrational wave functions
journal, November 2004
- Rauhut, Guntram
- The Journal of Chemical Physics, Vol. 121, Issue 19
On the use of quartic force fields in variational calculations
journal, June 2013
- Fortenberry, Ryan C.; Huang, Xinchuan; Yachmenev, Andrey
- Chemical Physics Letters, Vol. 574
Accurate calculation of vibrational frequencies using explicitly correlated coupled-cluster theory
journal, February 2009
- Rauhut, Guntram; Knizia, Gerald; Werner, Hans-Joachim
- The Journal of Chemical Physics, Vol. 130, Issue 5
High resolution infrared study of D2CO in the region of 1780–2400cm−1: assignment and preliminary analysis
journal, January 2006
- Lohilahti, J.; Ulenikov, O. N.; Bekhtereva, E. S.
- Journal of Molecular Structure, Vol. 780-781
The ν2 Band of Formaldehyde-d2
journal, February 2001
- Lohilahti, J.; Alanko, S.
- Journal of Molecular Spectroscopy, Vol. 205, Issue 2
New High-Resolution Analysis of the ν3, ν4, and ν6Bands of D2CO Measured by Fourier Transform Spectroscopy
journal, January 1998
- Perrin, A.; Flaud, J. -M.; Predoi-Cross, A.
- Journal of Molecular Spectroscopy, Vol. 187, Issue 1
Dispersed fluorescence spectroscopy of S0 vibrational levels in formaldehyde-d
journal, December 2008
- Ellsworth, Kristin K.; Lajiness, Brian D.; Lajiness, James P.
- Journal of Molecular Spectroscopy, Vol. 252, Issue 2
Submillimeter spectroscopy of H2C17O and a revisit of the rotational spectra of H2C18O and H2C16O
journal, January 2017
- Müller, Holger S. P.; Lewen, Frank
- Journal of Molecular Spectroscopy, Vol. 331
The rotational levels of the ground vibrational state of formaldehyde
journal, April 1997
- Carter, Stuart; Handy, Nicholas; Demaison, Jean
- Molecular Physics, Vol. 90, Issue 5
Works referencing / citing this record:
Laboratory spectroscopic study of isotopic thioformaldehyde, H 2 CS, and determination of its equilibrium structure
journal, January 2019
- Müller, Holger S. P.; Maeda, Atsuko; Thorwirth, Sven
- Astronomy & Astrophysics, Vol. 621
Vibrational analysis of the ubiquitous interstellar molecule cyclopropenylidene ( c -C 3 H 2 ): the importance of numerical stability
journal, March 2019
- Morgan, W. James; Fortenberry, Ryan C.; Schaefer III, Henry F.
- Molecular Physics, Vol. 118, Issue 1
Fourth-order vibrational perturbation theory with the Watson Hamiltonian: Report of working equations and preliminary results
journal, September 2018
- Gong, Justin Z.; Matthews, Devin A.; Changala, P. Bryan
- The Journal of Chemical Physics, Vol. 149, Issue 11
State‐of‐the‐art computations of dipole moments using analytic gradients of high‐level density‐fitted coupled‐cluster methods with focal‐point approximations
journal, December 2019
- Bozkaya, Uğur; Soydaş, Emine; Filiz, Bahar
- Journal of Computational Chemistry, Vol. 41, Issue 8
Theoretical studies of atmospheric molecular complexes interacting with NIR to UV light
journal, January 2018
- Biczysko, Malgorzata; Krupa, Justyna; Wierzejewska, Maria
- Faraday Discussions, Vol. 212
Astrophysical sulfur in diffuse and dark clouds: The fundamental vibrational frequencies and spectroscopic constants of hydrogen sulfide cation (H2S+)
journal, August 2018
- Morgan, W. James; Huang, Xinchuan; Schaefer, Henry F.
- Monthly Notices of the Royal Astronomical Society, Vol. 480, Issue 3