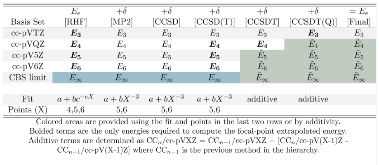
|
An accurate quartic force field for formaldehyde
|
journal
|
January 1996 |
|
The vibrations of formaldehyde
|
journal
|
July 1995 |
|
The Geometry of Formaldehyde
|
journal
|
September 1996 |
|
A new “spectroscopic” potential energy surface for formaldehyde in its ground electronic state
|
journal
|
June 2011 |
|
High-Accuracy ab Initio Rotation-Vibration Transitions for Water
|
journal
|
January 2003 |
|
On equilibrium structures of the water molecule
|
journal
|
June 2005 |
|
CVRQD ab initio ground-state adiabatic potential energy surfaces for the water molecule
|
journal
|
November 2006 |
|
Performance of W4 theory for spectroscopic constants and electrical properties of small molecules
|
journal
|
October 2010 |
|
A global potential energy surface and dipole moment surface for silane
|
journal
|
December 2015 |
|
Calculation of rotation-vibration energy levels of the ammonia molecule based on an ab initio potential energy surface
|
journal
|
September 2016 |
|
Full-Dimensional Potential Energy and Dipole Moment Surfaces of GeH 4 Molecule and Accurate First-Principle Rotationally Resolved Intensity Predictions in the Infrared
|
journal
|
November 2016 |
|
First fully ab initio potential energy surface of methane with a spectroscopic accuracy
|
journal
|
September 2016 |
|
In pursuit of the ab initio limit for conformational energy prototypes
|
journal
|
June 1998 |
|
Characterization of the X ̃ 1 A ’ state of isocyanic acid
|
journal
|
January 1993 |
|
The heat of formation of NCO
|
journal
|
September 1993 |
|
Revisitation of Nonorthogonal Spin Adaptation in Coupled Cluster Theory
|
journal
|
May 2013 |
|
The full CCSDT model for molecular electronic structure
|
journal
|
June 1987 |
|
Erratum: The full CCSDT model for molecular electronic structure [J. Chem. Phys. 8 6 , 7041 (1987)]
|
journal
|
September 1988 |
|
Coupled‐cluster method truncated at quadruples
|
journal
|
November 1991 |
|
The coupled‐cluster single, double, triple, and quadruple excitation method
|
journal
|
September 1992 |
|
Coupled-cluster methods including noniterative corrections for quadruple excitations
|
journal
|
August 2005 |
|
Non-orthogonal spin-adaptation of coupled cluster methods: A new implementation of methods including quadruple excitations
|
journal
|
February 2015 |
|
Accelerating the convergence of higher-order coupled cluster methods
|
journal
|
November 2015 |
|
Stimulated emission spectroscopy: A complete set of vibrational constants for X ̃ 1 A 1 formaldehyde
|
journal
|
June 1984 |
|
Stark level‐crossing spectroscopy of S 0 formaldehyde eigenstates at the dissociation threshold
|
journal
|
March 1990 |
|
Eigenstate‐resolved unimolecular reaction dynamics: Ergodic character of S 0 formaldehyde at the dissociation threshold
|
journal
|
March 1990 |
|
Pure vibrational spectroscopy of S 0 formaldehyde by dispersed fluorescence
|
journal
|
January 1996 |
|
High accuracy measurements on the ground state rotational spectrum of formaldehyde (H2CO) up to 2 THzElectronic supplementary information (ESI) available: Newly measured pure rotational transitions. See http://www.rsc.org/suppdata/cp/b3/b301657a/
|
journal
|
March 2003 |
|
FTFIR-spectrum of the ground state of D2CO
|
journal
|
November 2004 |
|
The HITRAN2016 molecular spectroscopic database
|
journal
|
December 2017 |
|
Spectroscopy of the formaldehyde isotopomer H213CO in the microwave to terahertz region
|
journal
|
January 2000 |
|
The accuracy of rotational constants predicted by high-level quantum-chemical calculations. I. molecules containing first-row atoms
|
journal
|
May 2008 |
|
The ab initio limit quartic force field of BH 3
|
journal
|
August 2005 |
|
A procedure for computing accurate ab initio quartic force fields: Application to HO2+ and H2O
|
journal
|
July 2008 |
|
Accurate ab initio quartic force fields for NH[sub 2]−] and CCH[sup −] and rovibrational spectroscopic constants for their isotopologs
|
journal
|
January 2009 |
|
The trans -HOCO radical: Quartic force fields, vibrational frequencies, and spectroscopic constants
|
journal
|
October 2011 |
|
Quartic force fields for excited electronic states: Rovibronic reference data for the 1 2A′ and 1 2A″ states of the isoformyl radical, HOC
|
journal
|
January 2015 |
|
A general, recursive, and open-ended response code
|
journal
|
February 2014 |
|
Benchmark calculations with correlated molecular wave functions. VII. Binding energy and structure of the HF dimer
|
journal
|
February 1995 |
|
Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions
|
journal
|
May 1992 |
|
Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited
|
journal
|
June 2001 |
|
The use of systematic sequences of wave functions for estimating the complete basis set, full configuration interaction limit in water
|
journal
|
May 1993 |
|
Basis-set convergence of correlated calculations on water
|
journal
|
June 1997 |
|
Importance of Angular Correlations between Atomic Electrons
|
journal
|
May 1962 |
|
Rates of convergence of the partial‐wave expansions of atomic correlation energies
|
journal
|
March 1992 |
|
Erratum: Rates of convergence of the partial‐wave expansions of atomic correlation energies [J. Chem. Phys. 96 , 4484 (1992)]
|
journal
|
December 1992 |
|
On the effectiveness of CCSD(T) complete basis set extrapolations for atomization energies
|
journal
|
July 2011 |
|
The atomization energy and proton affinity of NH3. An ab initio calibration study
|
journal
|
August 1996 |
|
The extrapolation of one-electron basis sets in electronic structure calculations: How it should work and how it can be made to work
|
journal
|
January 2005 |
|
HEAT: High accuracy extrapolated ab initio thermochemistry
|
journal
|
December 2004 |
|
High-accuracy extrapolated ab initio thermochemistry. II. Minor improvements to the protocol and a vital simplification
|
journal
|
August 2006 |
|
High-accuracy extrapolated ab initio thermochemistry. III. Additional improvements and overview
|
journal
|
March 2008 |
Psi4: an open-source ab initio electronic structure program : Psi4: an electronic structure program
- Turney, Justin M.; Simmonett, Andrew C.; Parrish, Robert M.
-
Wiley Interdisciplinary Reviews: Computational Molecular Science, Vol. 2, Issue 4
https://doi.org/10.1002/wcms.93
|
journal
|
October 2011 |
|
Approximate relativistic corrections to atomic radial wave functions*
|
journal
|
January 1976 |
|
Analytic second derivatives in high-order many-body perturbation and coupled-cluster theories: Computational considerations and applications
|
journal
|
January 2000 |
|
Analytic evaluation of the dipole Hessian matrix in coupled-cluster theory
|
journal
|
October 2013 |
|
Molecular equilibrium geometries based on coupled-cluster calculations including quadruple excitations
|
journal
|
August 2005 |
|
Basis-set extrapolation techniques for the accurate calculation of molecular equilibrium geometries using coupled-cluster theory
|
journal
|
July 2006 |
|
An Accurate ab Initio Quartic Force Field for Formaldehyde and Its Isotopomers
|
journal
|
July 1993 |
|
Calibration-quality adiabatic potential energy surfaces for H3+ and its isotopologues
|
journal
|
May 2012 |
Spectroscopy of H 3 + based on a new high-accuracy global potential energy surface
- Polyansky, Oleg L.; Alijah, Alexander; Zobov, Nikolai F.
-
Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, Vol. 370, Issue 1978
https://doi.org/10.1098/rsta.2012.0014
|
journal
|
November 2012 |
|
An accurate quartic force field and vibrational frequencies for HNO and DNO
|
journal
|
October 1994 |
|
General derivative relations for anharmonic force fields
|
journal
|
December 1996 |
|
A density matrix-based quasienergy formulation of the Kohn–Sham density functional response theory using perturbation- and time-dependent basis sets
|
journal
|
December 2008 |
|
The Dalton quantum chemistry program system: The Dalton program
|
journal
|
September 2013 |
|
GEN1INT: A unified procedure for the evaluation of one-electron integrals over Gaussian basis functions and their geometric derivatives
|
journal
|
August 2010 |
|
A unified scheme for the calculation of differentiated and undifferentiated molecular integrals over solid-harmonic Gaussians
|
journal
|
January 2007 |
|
Linear response calculations for large scale multiconfiguration self‐consistent field wave functions
|
journal
|
September 1988 |
|
Efficient elimination of response parameters in molecular property calculations for variational and nonvariational energies
|
journal
|
December 2008 |
|
Higher Order Rotation‐Vibration Energies of Polyatomic Molecules. IV
|
journal
|
September 1958 |
|
Theoretical studies of vibrationally excited polyatomic molecules using canonical Van Vleck perturbation theory
|
journal
|
April 1988 |
|
Numerical-Analytic Implementation of the Higher-Order Canonical Van Vleck Perturbation Theory for the Interpretation of Medium-Sized Molecule Vibrational Spectra
|
journal
|
March 2012 |
|
The prediction of molecular equilibrium structures by the standard electronic wave functions
|
journal
|
April 1997 |
|
The CO molecule: the role of basis set and correlation treatment in the calculation of molecular properties
|
journal
|
July 1997 |
|
Molecular Geometries at Sixth Order Møller−Plesset Perturbation Theory. At What Order Does MP Theory Give Exact Geometries?
|
journal
|
August 2000 |
|
The accurate determination of molecular equilibrium structures
|
journal
|
April 2001 |
|
Exact geometries from quantum chemical calculations
|
journal
|
June 2001 |
|
A priori calculation of molecular properties to chemical accuracy
|
journal
|
August 2004 |
|
CCSDT calculations of molecular equilibrium geometries
|
journal
|
August 1997 |
|
Communication: The performance of non-iterative coupled cluster quadruples models
|
journal
|
July 2015 |
|
Experimental, semi-experimental and ab initio equilibrium structures
|
journal
|
December 2007 |
|
Magnetic interactions in molecules and an analysis of molecular electronic charge distribution from magnetic parameters
|
journal
|
December 1974 |
|
The Molecular Zeeman Effect of Imines. I. Methanimine, its Molecular g-Tensor, its Magnetic Susceptibility Anisotropies, its Molecular Electric Quadrupole Moment, its Electric Field Gradient at the Nitrogen Nucleus, and its Nitrogen Spin-Rotation Coupling
|
journal
|
November 1989 |
|
Submillimeterwave spectrum of CH2PH and equilibrium structures of CH2PH and CH2NH
|
journal
|
August 2006 |
|
Basis set convergence for geometry and harmonic frequencies. Are h functions enough?
|
journal
|
August 1994 |
|
On the effect of core correlation on the geometry and harmonic frequencies of small polyatomic molecules
|
journal
|
August 1995 |
|
Basis Set Limit CCSD(T) Harmonic Vibrational Frequencies †
|
journal
|
November 2007 |
|
Coupled-cluster connected quadruples and quintuples corrections to the harmonic vibrational frequencies and equilibrium bond distances of HF, N2, F2, and CO
|
journal
|
September 2004 |
|
Factors Contributing to the Accuracy of Harmonic Force Field Calculations for Water
|
journal
|
May 2007 |
|
Analytic second derivatives for general coupled-cluster and configuration-interaction models
|
journal
|
April 2004 |
|
The geometry, vibrational frequencies, and total atomization energy of ethylene. A calibration study
|
journal
|
January 1996 |
|
Ab Initio Calibration Study of the Heat of Formation, Geometry, and Anharmonic Force Field of Fluoroacetylene
|
journal
|
March 1998 |
|
High level coupled cluster determination of the structure, frequencies, and heat of formation of water
|
journal
|
October 2009 |
|
Anharmonic vibrational analysis of water with traditional and explicitly correlated coupled cluster methods
|
journal
|
August 2010 |
|
A systematic study of molecular vibrational anharmonicity and vibration—rotation interaction by self-consistent-field higher-derivative methods. Asymmetric top molecules
|
journal
|
July 1988 |
|
The Vibration-Rotation Energies of Polyatomic Molecules Part II. Accidental Degeneracies
|
journal
|
October 1945 |
|
The anharmonic force field of ethylene, C 2 H 4 , by means of accurate ab initio calculations
|
journal
|
August 1995 |
|
Calculated stretching overtone levels and Darling–Dennison resonances in water: a triumph of simple theoretical approaches
|
journal
|
October 2007 |
|
Quantitative analysis of Fermi resonances by harmonic derivatives of perturbation theory corrections
|
journal
|
October 2009 |
|
Beyond the x - K relations : Calculations of 1–1 and 2–2 resonance constants with application to HCN and DCN
|
journal
|
April 1989 |
|
Accurate ab initio quartic force field for trans-HNNH and treatment of resonance polyads
|
journal
|
July 1997 |
|
VPT2+K spectroscopic constants and matrix elements of the transformed vibrational Hamiltonian of a polyatomic molecule with resonances using Van Vleck perturbation theory
|
journal
|
June 2013 |
|
Calculation of fundamental frequencies for small polyatomic molecules: a comparison between correlation consistent and atomic natural orbital basis sets
|
journal
|
May 2013 |
|
New analysis of the ν2 band of formaldehyde : Line positions for the ν2, ν3, ν4 and ν6 interacting bands
|
journal
|
October 2007 |
|
New analysis of the 2ν4, ν4+ν6, 2ν6, ν3+ν4, ν3+ν6, ν1, ν5, ν2+ν4, 2ν3, ν2+ν6 and ν2+ν3 bands of formaldehyde H212C16O: Line positions and intensities in the 3.5μm spectral region
|
journal
|
January 2006 |
|
High resolution spectroscopy of H 2 12 C 16 O in the 1.9 to 2.56 µm spectral range
|
journal
|
June 2006 |
|
Efficient calculation of potential energy surfaces for the generation of vibrational wave functions
|
journal
|
November 2004 |
|
On the use of quartic force fields in variational calculations
|
journal
|
June 2013 |
|
Accurate calculation of vibrational frequencies using explicitly correlated coupled-cluster theory
|
journal
|
February 2009 |
|
High resolution infrared study of D2CO in the region of 1780–2400cm−1: assignment and preliminary analysis
|
journal
|
January 2006 |
|
The ν2 Band of Formaldehyde-d2
|
journal
|
February 2001 |
|
New High-Resolution Analysis of the ν3, ν4, and ν6Bands of D2CO Measured by Fourier Transform Spectroscopy
|
journal
|
January 1998 |
|
Dispersed fluorescence spectroscopy of S0 vibrational levels in formaldehyde-d
|
journal
|
December 2008 |
|
Submillimeter spectroscopy of H2C17O and a revisit of the rotational spectra of H2C18O and H2C16O
|
journal
|
January 2017 |
|
The rotational levels of the ground vibrational state of formaldehyde
|
journal
|
April 1997 |