
Size-dependent error of the density functional theory ionization potential in vacuum and solution
Abstract
Density functional theory is often the method of choice for modeling the energetics of large molecules and including explicit solvation effects. It is preferable to use a method that treats systems of different sizes and with different amounts of explicit solvent on equal footing. However, recent work suggests that approximate density functional theory has a size-dependent error in the computation of the ionization potential. We here investigate the lack of size-intensivity of the ionization potential computed with approximate density functionals in vacuum and solution. We show that local and semi-local approximations to exchange do not yield a constant ionization potential for an increasing number of identical isolated molecules in vacuum. Instead, as the number of molecules increases, the total energy required to ionize the system decreases. Rather surprisingly, we find that this is still the case in solution, whether using a polarizable continuum model or with explicit solvent that breaks the degeneracy of each solute, and we find that explicit solvent in the calculation can exacerbate the size-dependent delocalization error. We demonstrate that increasing the amount of exact exchange changes the character of the polarization of the solvent molecules; for small amounts of exact exchange the solvent molecules contribute amore »
- Authors:
-
- Univ. of California, Merced, CA (United States)
- Publication Date:
- Research Org.:
- Univ. of California, Merced, CA (United States)
- Sponsoring Org.:
- USDOE Office of Science (SC), Basic Energy Sciences (BES)
- OSTI Identifier:
- 1233558
- Alternate Identifier(s):
- OSTI ID: 1234092
- Grant/Contract Number:
- SC0014437
- Resource Type:
- Accepted Manuscript
- Journal Name:
- Journal of Chemical Physics
- Additional Journal Information:
- Journal Volume: 143; Journal Issue: 24; Journal ID: ISSN 0021-9606
- Publisher:
- American Institute of Physics (AIP)
- Country of Publication:
- United States
- Language:
- English
- Subject:
- 71 CLASSICAL AND QUANTUM MECHANICS, GENERAL PHYSICS; density functional theory; solvents; ionization; exact exchange; size-dependent error; polarization; single molecule techniques
Citation Formats
Sosa Vazquez, Xochitl A., and Isborn, Christine M. Size-dependent error of the density functional theory ionization potential in vacuum and solution. United States: N. p., 2015.
Web. doi:10.1063/1.4937417.
Sosa Vazquez, Xochitl A., & Isborn, Christine M. Size-dependent error of the density functional theory ionization potential in vacuum and solution. United States. https://doi.org/10.1063/1.4937417
Sosa Vazquez, Xochitl A., and Isborn, Christine M. Tue .
"Size-dependent error of the density functional theory ionization potential in vacuum and solution". United States. https://doi.org/10.1063/1.4937417. https://www.osti.gov/servlets/purl/1233558.
@article{osti_1233558,
title = {Size-dependent error of the density functional theory ionization potential in vacuum and solution},
author = {Sosa Vazquez, Xochitl A. and Isborn, Christine M.},
abstractNote = {Density functional theory is often the method of choice for modeling the energetics of large molecules and including explicit solvation effects. It is preferable to use a method that treats systems of different sizes and with different amounts of explicit solvent on equal footing. However, recent work suggests that approximate density functional theory has a size-dependent error in the computation of the ionization potential. We here investigate the lack of size-intensivity of the ionization potential computed with approximate density functionals in vacuum and solution. We show that local and semi-local approximations to exchange do not yield a constant ionization potential for an increasing number of identical isolated molecules in vacuum. Instead, as the number of molecules increases, the total energy required to ionize the system decreases. Rather surprisingly, we find that this is still the case in solution, whether using a polarizable continuum model or with explicit solvent that breaks the degeneracy of each solute, and we find that explicit solvent in the calculation can exacerbate the size-dependent delocalization error. We demonstrate that increasing the amount of exact exchange changes the character of the polarization of the solvent molecules; for small amounts of exact exchange the solvent molecules contribute a fraction of their electron density to the ionized electron, but for larger amounts of exact exchange they properly polarize in response to the cationic solute. As a result, in vacuum and explicit solvent, the ionization potential can be made size-intensive by optimally tuning a long-range corrected hybrid functional.},
doi = {10.1063/1.4937417},
journal = {Journal of Chemical Physics},
number = 24,
volume = 143,
place = {United States},
year = {Tue Dec 22 00:00:00 EST 2015},
month = {Tue Dec 22 00:00:00 EST 2015}
}
 Search WorldCat to find libraries that may hold this journal
Search WorldCat to find libraries that may hold this journalWeb of Science
Figures / Tables:
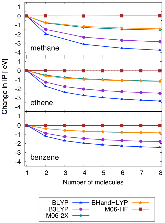 FIG. 1: Change in DFT computed ionization potential (IP) compared to a single molecule for increasing number of noninteracting methane, ethene, and benzene molecules in vacuum.
FIG. 1: Change in DFT computed ionization potential (IP) compared to a single molecule for increasing number of noninteracting methane, ethene, and benzene molecules in vacuum.
Works referenced in this record:
Quantum Chemistry on Graphical Processing Units. 2. Direct Self-Consistent-Field Implementation
journal, March 2009
- Ufimtsev, Ivan S.; Martinez, Todd J.
- Journal of Chemical Theory and Computation, Vol. 5, Issue 4
Excited-State Electronic Structure with Configuration Interaction Singles and Tamm–Dancoff Time-Dependent Density Functional Theory on Graphical Processing Units
journal, May 2011
- Isborn, Christine M.; Luehr, Nathan; Ufimtsev, Ivan S.
- Journal of Chemical Theory and Computation, Vol. 7, Issue 6
Quantum Chemical Calculations Using Accelerators: Migrating Matrix Operations to the NVIDIA Kepler GPU and the Intel Xeon Phi
journal, February 2014
- Leang, Sarom S.; Rendell, Alistair P.; Gordon, Mark S.
- Journal of Chemical Theory and Computation, Vol. 10, Issue 3
Computation of the Density Matrix in Electronic Structure Theory in Parallel on Multiple Graphics Processing Units
journal, November 2014
- Cawkwell, M. J.; Wood, M. A.; Niklasson, Anders M. N.
- Journal of Chemical Theory and Computation, Vol. 10, Issue 12
Relationship between long-range charge-transfer excitation energy error and integer discontinuity in Kohn–Sham theory
journal, December 2003
- Tozer, David J.
- The Journal of Chemical Physics, Vol. 119, Issue 24
Fundamental gaps with approximate density functionals: The derivative discontinuity revealed from ensemble considerations
journal, May 2014
- Kraisler, Eli; Kronik, Leeor
- The Journal of Chemical Physics, Vol. 140, Issue 18
A self-interaction-free local hybrid functional: Accurate binding energies vis-à-vis accurate ionization potentials from Kohn-Sham eigenvalues
journal, May 2014
- Schmidt, Tobias; Kraisler, Eli; Makmal, Adi
- The Journal of Chemical Physics, Vol. 140, Issue 18
A challenge for density functionals: Self-interaction error increases for systems with a noninteger number of electrons
journal, August 1998
- Zhang, Yingkai; Yang, Weitao
- The Journal of Chemical Physics, Vol. 109, Issue 7
Many-electron self-interaction error in approximate density functionals
journal, November 2006
- Mori-Sánchez, Paula; Cohen, Aron J.; Yang, Weitao
- The Journal of Chemical Physics, Vol. 125, Issue 20
Insights into Current Limitations of Density Functional Theory
journal, August 2008
- Cohen, A. J.; Mori-Sanchez, P.; Yang, W.
- Science, Vol. 321, Issue 5890
Structural manifestation of the delocalization error of density functional approximations: C4N+2 rings and C20 bowl, cage, and ring isomers
journal, June 2010
- Heaton-Burgess, Tim; Yang, Weitao
- The Journal of Chemical Physics, Vol. 132, Issue 23
Delocalization error of density-functional approximations: A distinct manifestation in hydrogen molecular chains
journal, December 2012
- Zheng, Xiao; Liu, Min; Johnson, Erin R.
- The Journal of Chemical Physics, Vol. 137, Issue 21
Extreme density-driven delocalization error for a model solvated-electron system
journal, November 2013
- Johnson, Erin R.; Otero-de-la-Roza, A.; Dale, Stephen G.
- The Journal of Chemical Physics, Vol. 139, Issue 18
Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy
journal, December 1982
- Perdew, John P.; Parr, Robert G.; Levy, Mel
- Physical Review Letters, Vol. 49, Issue 23
Tests of functionals for systems with fractional electron number
journal, April 2007
- Vydrov, Oleg A.; Scuseria, Gustavo E.; Perdew, John P.
- The Journal of Chemical Physics, Vol. 126, Issue 15
Fractional charge perspective on the band gap in density-functional theory
journal, March 2008
- Cohen, Aron J.; Mori-Sánchez, Paula; Yang, Weitao
- Physical Review B, Vol. 77, Issue 11
Curvature and Frontier Orbital Energies in Density Functional Theory
journal, December 2012
- Stein, Tamar; Autschbach, Jochen; Govind, Niranjan
- The Journal of Physical Chemistry Letters, Vol. 3, Issue 24
Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange
journal, August 2003
- Dreuw, Andreas; Weisman, Jennifer L.; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 119, Issue 6
A long-range-corrected time-dependent density functional theory
journal, May 2004
- Tawada, Yoshihiro; Tsuneda, Takao; Yanagisawa, Susumu
- The Journal of Chemical Physics, Vol. 120, Issue 18
Kohn-Sham Self-Interaction Correction in Real Time
journal, April 2012
- Hofmann, D.; Körzdörfer, T.; Kümmel, S.
- Physical Review Letters, Vol. 108, Issue 14
Generalized Kohn-Sham schemes and the band-gap problem
journal, February 1996
- Seidl, A.; Görling, A.; Vogl, P.
- Physical Review B, Vol. 53, Issue 7
Coulomb-attenuated exchange energy density functionals
journal, July 1996
- Gill, Peter M. W.; Adamson, Ross D.; Pople, John A.
- Molecular Physics, Vol. 88, Issue 4
A long-range correction scheme for generalized-gradient-approximation exchange functionals
journal, August 2001
- Iikura, Hisayoshi; Tsuneda, Takao; Yanai, Takeshi
- The Journal of Chemical Physics, Vol. 115, Issue 8
A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP)
journal, July 2004
- Yanai, Takeshi; Tew, David P.; Handy, Nicholas C.
- Chemical Physics Letters, Vol. 393, Issue 1-3, p. 51-57
Long-range–short-range separation of the electron-electron interaction in density-functional theory
journal, December 2004
- Toulouse, Julien; Colonna, François; Savin, Andreas
- Physical Review A, Vol. 70, Issue 6
Density Functional Theory with Correct Long-Range Asymptotic Behavior
journal, February 2005
- Baer, Roi; Neuhauser, Daniel
- Physical Review Letters, Vol. 94, Issue 4
Systematic optimization of long-range corrected hybrid density functionals
journal, February 2008
- Chai, Jeng-Da; Head-Gordon, Martin
- The Journal of Chemical Physics, Vol. 128, Issue 8
Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals
journal, April 2012
- Kronik, Leeor; Stein, Tamar; Refaely-Abramson, Sivan
- Journal of Chemical Theory and Computation, Vol. 8, Issue 5
Delocalization Error and “Functional Tuning” in Kohn–Sham Calculations of Molecular Properties
journal, June 2014
- Autschbach, Jochen; Srebro, Monika
- Accounts of Chemical Research, Vol. 47, Issue 8
Organic Electronic Materials: Recent Advances in the DFT Description of the Ground and Excited States Using Tuned Range-Separated Hybrid Functionals
journal, April 2014
- Körzdörfer, Thomas; Brédas, Jean-Luc
- Accounts of Chemical Research, Vol. 47, Issue 11
Prediction of charge-transfer excitations in coumarin-based dyes using a range-separated functional tuned from first principles
journal, December 2009
- Stein, Tamar; Kronik, Leeor; Baer, Roi
- The Journal of Chemical Physics, Vol. 131, Issue 24
Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory
journal, March 2009
- Stein, Tamar; Kronik, Leeor; Baer, Roi
- Journal of the American Chemical Society, Vol. 131, Issue 8
Koopmans’ springs to life
journal, December 2009
- Salzner, Ulrike; Baer, Roi
- The Journal of Chemical Physics, Vol. 131, Issue 23
Tuned Range-Separated Hybrids in Density Functional Theory
journal, March 2010
- Baer, Roi; Livshits, Ester; Salzner, Ulrike
- Annual Review of Physical Chemistry, Vol. 61, Issue 1
Charge-Transfer-Like π→π* Excitations in Time-Dependent Density Functional Theory: A Conundrum and Its Solution
journal, July 2011
- Kuritz, Natalia; Stein, Tamar; Baer, Roi
- Journal of Chemical Theory and Computation, Vol. 7, Issue 8
Quasiparticle Spectra from a Nonempirical Optimally Tuned Range-Separated Hybrid Density Functional
journal, November 2012
- Refaely-Abramson, Sivan; Sharifzadeh, Sahar; Govind, Niranjan
- Physical Review Letters, Vol. 109, Issue 22
Optimum Exchange for Calculation of Excitation Energies and Hyperpolarizabilities of Organic Electro-optic Chromophores
journal, August 2014
- Garrett, Kerry; Sosa Vazquez, XochitlA; Egri, Shawn B.
- Journal of Chemical Theory and Computation, Vol. 10, Issue 9
Improved Prediction of Properties of π-Conjugated Oligomers with Range-Separated Hybrid Density Functionals
journal, July 2011
- Salzner, Ulrike; Aydin, Aykut
- Journal of Chemical Theory and Computation, Vol. 7, Issue 8
Influence of the Delocalization Error and Applicability of Optimal Functional Tuning in Density Functional Calculations of Nonlinear Optical Properties of Organic Donor-Acceptor Chromophores
journal, June 2013
- Sun, Haitao; Autschbach, Jochen
- ChemPhysChem, Vol. 14, Issue 11
Density-functional errors in ionization potential with increasing system size
journal, May 2015
- Whittleton, Sarah R.; Sosa Vazquez, Xochitl A.; Isborn, Christine M.
- The Journal of Chemical Physics, Vol. 142, Issue 18
Tuned range separated hybrid functionals for solvated low bandgap oligomers
journal, July 2015
- de Queiroz, Thiago B.; Kümmel, Stephan
- The Journal of Chemical Physics, Vol. 143, Issue 3
Counterintuitive electron localisation from density-functional theory with polarisable solvent models
journal, November 2015
- Dale, Stephen G.; Johnson, Erin R.
- The Journal of Chemical Physics, Vol. 143, Issue 18
Density-functional exchange-energy approximation with correct asymptotic behavior
journal, September 1988
- Becke, A. D.
- Physical Review A, Vol. 38, Issue 6
Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density
journal, January 1988
- Lee, Chengteh; Yang, Weitao; Parr, Robert G.
- Physical Review B, Vol. 37, Issue 2
Density‐functional thermochemistry. III. The role of exact exchange
journal, April 1993
- Becke, Axel D.
- The Journal of Chemical Physics, Vol. 98, Issue 7, p. 5648-5652
The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals
journal, July 2007
- Zhao, Yan; Truhlar, Donald G.
- Theoretical Chemistry Accounts, Vol. 120, Issue 1-3
Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States
journal, December 2006
- Zhao, Yan; Truhlar, Donald G.
- The Journal of Physical Chemistry A, Vol. 110, Issue 49
Comparison of simple potential functions for simulating liquid water
journal, July 1983
- Jorgensen, William L.; Chandrasekhar, Jayaraman; Madura, Jeffry D.
- The Journal of Chemical Physics, Vol. 79, Issue 2
Hartree-Fock instabilities and electronic properties
journal, April 2000
- Dehareng, Dominique; Dive, Georges
- Journal of Computational Chemistry, Vol. 21, Issue 6
Entanglement and Polyradical Character of Polycyclic Aromatic Hydrocarbons Predicted by Projected Hartree–Fock Theory
journal, May 2013
- Rivero, Pablo; Jiménez-Hoyos, Carlos A.; Scuseria, Gustavo E.
- The Journal of Physical Chemistry B, Vol. 117, Issue 42
Works referencing / citing this record:
Dielectric Screening Meets Optimally Tuned Density Functionals
journal, April 2018
- Kronik, Leeor; Kümmel, Stephan
- Advanced Materials, Vol. 30, Issue 41
Modeling absorption spectra of molecules in solution
journal, September 2018
- Zuehlsdorff, Tim J.; Isborn, Christine M.
- International Journal of Quantum Chemistry, Vol. 119, Issue 1
Noncentrosymmetric organic crystals of barbiturates as potential nonlinear optical phores: experimental and theoretical analyses
journal, June 2019
- Ivanova, Bojidarka; Spiteller, Michael
- Chemical Papers, Vol. 73, Issue 11
Density functional approximations for orbital energies and total energies of molecules and solids
journal, August 2018
- Baerends, Evert Jan
- The Journal of Chemical Physics, Vol. 149, Issue 5
On combining the conductor-like screening model and optimally tuned range-separated hybrid density functionals
journal, May 2019
- Sachse, Torsten; Martínez, Todd J.; Presselt, Martin
- The Journal of Chemical Physics, Vol. 150, Issue 17
Prediction of electronic couplings for molecular charge transfer using optimally tuned range-separated hybrid functionals
journal, March 2018
- Manna, Debashree; Blumberger, Jochen; Martin, Jan M. L.
- Molecular Physics, Vol. 116, Issue 19-20
Prediction of electronic couplings for molecular charge transfer using optimally tuned range-separated hybrid functionals
text, January 2020
- Manna, Debashree; Blumberger, Jochen; Martin, Jan M. L.
- Taylor & Francis
Prediction of electronic couplings for molecular charge transfer using optimally tuned range-separated hybrid functionals
text, January 2018
- Manna, Debashree; Blumberger, Jochen; Martin, Jan M. L.
- Taylor & Francis
Figures / Tables found in this record: